
[This commentary post is by Joe Pickrell on Finding the sources of missing heritability in a yeast cross by Bloom et al., available from the arXiv here]
For decades, human geneticists have used twin studies and related methods to show that a considerable amount of the phenotypic variation in humans is driven by genetic variation (without any knowledge of the underlying loci). More recently, genome-wide association studies have made incredible progress in identifying specific genetic variants that are important in various traits. However, the loci identified in the latter studies often have small effects, and the sums of their effects rarely come close to the genetic effects known from the former. The difference between the genetic effects on a trait known from heritability studies and the effects estimated from individual loci has come to be known as the “missing heritability”, and much ink has been spilled on speculation as to its cause.

Bloom et al. take an elegant and straightforward approach to this question using a model system, the budding yeast Saccharomyces cerevisiae. The insight is that in order to make progress, one needs both an experimental design to isolate some of the possible causes of the “missing heritability”. To achieve this, Bloom et al. use a cross between two different yeast strains and grow the segregants in identical conditions. They thus remove much of the environmental variation in the phenotypes, while also removing the effect of allele frequency (since all alleles are at frequency 0.5 in the cross). While this means that they cannot address some controversies about potential sources of heritability in humans (for example, rare versus common variants), they are able to estimate how much phenotypic variation is due to detectable additive effects. The authors additionally develop high-throughput assays to measure phenotypes (in their case, growth rates in 46 different conditions) and genotypes in the segregants from the cross, so that they can perform high-powered mapping studies.
The main result relevant to the issue of “missing heritability” is presented in their Figure 3a (reproduced above). After performing a well-powered mapping study, the authors compare the effects from their identified loci to the narrow-sense heritability of each trait. As it turns out, the heritability is not missing! In this cross, the identified loci, though of small effect, add up to a substantial fraction of the overall (narrow-sense) heritability for most traits. The authors additional identify some gene-gene interactions that contribute to the broad-sense heritability (but by definition not the narrow-sense heritability) of many traits.
The authors provocatively interpret their results as supporting a model in which the majority of the “missing heritability” lies in a large number of variants with small effect sizes (in line with the model proposed most notably by Peter Visscher and colleagues, though the authors here make no claims about the allele frequencies of the relevant variants). While this seems to be true in yeast, it remains to be seen in humans. It’s of course easy to come up with reasons why this might not hold in humans–our species is a special snowflake and so forth–but this paper should be in the back of the mind of anyone who is thinking about this problem.

I am a little unclear about the differences between narrow and broad heritability. Could you give me an example of one or the other?
I get a little turned around thinking about this sometimes as well. Here’s a non-traditional sort of example that I think is correct:
Imagine you’re trying to predict my height using my genotype data and imagine you know every relevant site in the genome. You build an additive model such that if I have one copy of a height increasing allele at a given site you add to your prediction and if I have two copies you add
to your prediction and if I have two copies you add  to your prediction. The best you can do at predicting my height is limited by the narrow-sense heritability.
to your prediction. The best you can do at predicting my height is limited by the narrow-sense heritability.
Now consider a more complex model, where you add dominance terms, such that if I have two copies of a height-increasing allele you no longer add to my height, but instead you add
to my height, but instead you add 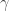 . Additionally, you model interactions between loci, such that the effect on my height at locus 1 is influenced by my genotype at locus 2. If you knew all these parameters, the best you could do at predicting my height is limited by the broad-sense heritability.
. Additionally, you model interactions between loci, such that the effect on my height at locus 1 is influenced by my genotype at locus 2. If you knew all these parameters, the best you could do at predicting my height is limited by the broad-sense heritability.
So narrow-sense is variance due to additive genetic effects only, broad-sense includes other sorts of things. Others reading this, feel free to correct me on the details here.
To non-biologists (usually physical scientists) I just say that:
* Heritability is proportion of trait variation due to genetic variation
* Additive variance ~ linear genetic effects
* Narrow sense heritability is proportion of trait variation due to additive variance
* Broad sense includes non-linear genetic effects (dominance, interaction, etc.)
Is the emphasis I make on linear vs. non-linear incorrect out of curiosity?
Pingback: Thinking about heritability | Gene Expression | Discover Magazine
Pingback: Thinking about heritability | Biology News by Biologged
Pingback: Distributing the origins of human will | Gene Expression | Discover Magazine
Pingback: Distributing the origins of human will | Biology News by Biologged
Pingback: Most viewed on Haldane’s Sieve: October 2012 | Haldane's Sieve
Pingback: Most viewed on Haldane’s Sieve: December 2012 | Haldane's Sieve
Pingback: Haldane’s Sieve sifts through 2012 | Haldane's Sieve
Pingback: Where’s the heritability? Right where you’d expect—if you look close enough | The Molecular Ecologist
Pingback: Yeast Genomics Helps Explaining Missing Heritability