
This post is by Graham Coop [@Graham_Coop], and is an introduction to the background of Anand Bhaskar and Yun Song preprint: “The identifiability of piecewise demographic models from the sample frequency spectrum”. arXived here.
Anand and Yun’s preprint focuses on what we can learn about population demographic history from the site frequency spectrum. It takes as its starting point an article by Myers et al (Myers, S., Fefferman, C., and Patterson, N. (2008) Can one learn history from the allelic spectrum? Theoretical Population Biology 73, 342–348.). I wrote about Myers et al’s article back in 2008, when it came out (see here). I thought I’d post an edited version of that post by way of additional background to Anand and Yun’s preprint and guest post.
Edited version of original post:
The best way to learn about demography from population genetic data is to look at patterns of diversity across many unlinked regions. The distribution of frequencies in a populations of unlinked neutral alleles at SNPs (the site frequency spectrum) is potentially very informative about population history. For example an excess of low frequency mutations is consistent with recent population growth, as the increase in population size introduces new mutations but these mutations have not yet had time to drift to higher frequencies. Many authors have made use of the frequency spectrum of unlinked, putatively neutral SNPs to learn about demography.
Back in 2008 a technical but elegant article by Myers et al shows that while informative about demography, the site frequency spectrum at unlinked SNPs can not help you chose between certain demographic histories. This is not a question of imperfect knowledge of the site frequency spectrum (which more data would solve) but because for any particular demographic model, as Myers et al formally show, there are a large family of demographic histories that can give rise to the same site frequency spectrum. They explained: ‘Informally, changes in population size at some past time are canceled out by other changes in the opposite direction’. I think that this lack of information comes from the fact that each unlinked SNP only tells you about the placement of a single mutation on the genealogy of the population at that site, and over sites you learn about the expected amount of time in different parts of the genealogy. By fluctuating the population size in just the right way, i.e. speeding up and slowing down the rate of coalescence, we can get very different population histories to give us the same expected coalescent times.
For example Myers et al showed that the population size history below, gives the same population frequency spectrum as a a constant population
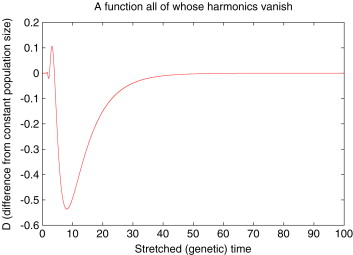
Figure taken from Myers, S., Fefferman, C., and Patterson, N. (2008) Can one learn history from the allelic spectrum? Theoretical Population Biology 73, 342–348.
That’s somewhat worrying as it says that when we fit a model of population size changes over time there are actually a family of quite different looking population histories that would give us just as good a fit to the data. However, these alternative histories do look quite strange and may not be biologically reasonable.
Anand and Yun’s article takes this as their starting place. They show that if we write our population history as a series of piecewise functions that the parameters of these functions are identifable, and provide simple estimates of the sample size needed. You can read more about their results in their guest post here at Haldane’s sieve.

Pingback: Most viewed on Haldane’s Sieve: September 2013 | Haldane's Sieve