Horizontal gene transfer may explain variation in θs
Rohan Maddamsetti, Philip J. Hatcher, Stéphane Cruveiller, Claudine Médigue, Jeffrey E. Barrick, Richard E. Lenski
(Submitted on 28 Sep 2012)
Martincorena et al. estimated synonymous diversity ( ) across 2,930 orthologous gene alignments from 34 Escherichia coli genomes, and found substantial variation among genes in the density of synonymous polymorphisms. They argue that this pattern reflects variation in the mutation rate per nucleotide (
) across 2,930 orthologous gene alignments from 34 Escherichia coli genomes, and found substantial variation among genes in the density of synonymous polymorphisms. They argue that this pattern reflects variation in the mutation rate per nucleotide (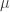 ) among genes. However, the effective population size (N) is not necessarily constant across the genome. In particular, different genes may have different histories of horizontal gene transfer (HGT), whereas Martincorena et al. used a model with random recombination to calculate
) among genes. However, the effective population size (N) is not necessarily constant across the genome. In particular, different genes may have different histories of horizontal gene transfer (HGT), whereas Martincorena et al. used a model with random recombination to calculate  . They did filter alignments in an effort to minimize the effects of HGT, but we doubt that any procedure can completely eliminate HGT among closely related genomes, such as E. coli living in the complex gut community.
. They did filter alignments in an effort to minimize the effects of HGT, but we doubt that any procedure can completely eliminate HGT among closely related genomes, such as E. coli living in the complex gut community.
Here we show that there is no significant variation among genes in rates of synonymous substitutions in a long-term evolution experiment with E. coli and that the per-gene rates are not correlated with estimates from genome comparisons. However, there is a significant association between and HGT events. Together, these findings imply that variation reflects different histories of HGT, not local optimization of mutation rates to reduce the risk of deleterious mutations as proposed by Martincorena et al.


A minor comment on what seems like a solid response. The decomposition of synonymous diversity into the product of the effective population size and mutation rate isn’t always the most straight forward way to discuss levels.of diversity at putatively neutral sites. It might be better to say that neutral synonymous diversity reflects 2 \mu T, where T is the time to the common ancestor of a pair of sequences. In that setting it is obvious that HGT leads to wide variation in T. I tend to only use the \mu Ne version where there is some hope that we can understand patterns of neutral diversity through an analogy to a constant population with rescaled the rate of drift (Ne).
Unfortunately this is a very misleading paper with multiple erroneous claims:
1. Maddamsetti et al. claim to have found an association between thetaS and HGT. However this association is only present in their own calculations of thetaS, not in the calculations reported in Nature (Martincorena et al). This is not surprising as in the original paper HGT was carefully filtered by a series of population genetic and phylogenetic tests. Many details can be found in the Supplementary Information of Martincorena et al. http://www.nature.com/nature/journal/v485/n7396/extref/nature10995-s1.pdf. To make matters worse Maddamsetti et al. knew that their observation did not hold using the published estimates but did not acknowledge it.
2. Maddamsetti et al. are using a collection of SNPs found in repair-deficient strains with genomic mutation rates around two orders of magnitude higher than the wildtype and with a very unusual spectrum of mutations. In these conditions mutations are very likely to occur randomly hiding any possible non-randomness in the mutational processes that dominate the mutation spectrum of the wild-type. Thus, observations in these conditions cannot be used to prove or disprove observations that require wild-type conditions.
3. Finally, the only result provided by Maddamsetti et al., other than the invalid HGT analysis, is the use of a cumulative plot to demonstrate that their data fit a uniform model (number of mutations proportional to gene length) better than a heterogeneous model. Very unfortunately, this plot is also wrong. First, the heterogeneous model is expressed as mutations ~ thetaS, when it obviously should be expressed as mutations ~ thetaS*gene_length (given that thetaS is an estimate of mutation rate per base). Second, the cumulative plot implicitly expects the data to fit sampling variation present in the estimates of Martincorena et al. Third, a bit of playing with this representation reveals that one can simulate extremely variable mutation rates and the resulting highly heterogeneous mutations would still appear to fit the uniform model perfectly given the dominant effect of gene length and the fact that using a cumulative function hides any variation added by a single gene in each step.
It is very unfortunate that Maddamsetti et al. did not subject their publication to peer-review before posting this manuscript as it clearly does not meet a minimum quality standard. I recommend the readers to form their opinions after carefully reading the extensive supplementary material by Martincorena et al. There they will find extensive analyses on the role of HGT, within-species homologous recombination, selection on synonymous sites, hitch-hiking, background selection, etc. http://www.nature.com/nature/journal/v485/n7396/extref/nature10995-s1.pdf
RS, are you an author of Martincorena et al.? We encourage academics to comment under their real names, as it helps to maintain civility.
Sorry Joe, yes I am, I assumed it was clear, apologies for the confusion. I am actually the first author of the paper. We are hoping to respond to Maddamsetti et al. in a peer-reviewed context in a near future, if we are given the chance. In the meantime, however, I hope that the points mentioned here help the reader to form their own opinion. Thanks! -Inigo
Thanks for clarifying!
Hi everybody,
Titus Brown asked me to write a blog post on why this stuff is interesting; I’ll post the link when it’s up. I’ll respond to these technical criticisms on this blog; if anyone’s interested, my data and code is online here: http://code.google.com/p/luscombe-reanalysis/. I just added the file that Inigo sent me comparing my thetaS estimates to his to the repo (with minor formatting changes to make it play better with R– csv instead of tab-delimited, NA instead of NaN), so now everyone can compare our results.
Now for my technical comments:
cooplab, I agree with you that synonymous diversity is actually 2uT, and that 2u*Ne is when T is measured in Ne generations in the relevant Wright-Fisher process (I believe this is the coalescent effective pop. size, though? Correct me if I’m wrong.) Another way of looking at HGT though, is that some genes actually physically have a larger population size than other genes, say because they get transferred successfully more often, or because they’re generally found across more microbes. This still cashes out the same way, since larger pop. size means longer time to LCA, thus higher synonymous diversity. Different way of making the same point, in my mind.
As for Inigo’s comments, I do hope we get this exchange published in print in a more rigorous forum, but this still captures the essence of ‘peer-review’ right? I put my science up, other people throw rocks at it, and at the end we hopefully have a better understanding of how things work. Also, blogs don’t have word count limits 😉
1. I think it’s important to note that my association between thetaS and HGT was the product of a actual hypothesis test (are thetaS and HGT correlated? In my results, yes.) Inigo’s risk-management hypothesis, on the other hand is a conjecture that fits the data after the fact (nothing wrong with that, but it certainly hasn’t been tested). It is true that there’s no significant association between the HGT and Inigo’s thetaS estimates, but this is due to his filters for HGT! In my mind, this shows that HGT can indeed inflate thetaS. That said, it is certainly debatable whether HGT explains the rest of the reported variation in thetaS–Inigo makes the claim that there’s no HGT at ALL in his alignments due to his filters, but this is difficult to evaluate without calculating specificity and sensitivity for these filters based on some gold-standard HGT dataset. I also think that it’s important to note that I don’t claim to have demonstrated that HGT causes all observed variation in thetaS–I do believe that it’s an important part of the story, though. Obviously Inigo and I disagree on this. In a nutshell, I think that the number of genes that haven’t undergone HGT will approach zero as sampling within Inigo’s species tree of 34 E. coli increases, and that many HGT events have occurred along each branch that simply can’t be detected from only looking at 34 E. coli genomes (and this is ignoring E. coli that are outgroups to these 34 genomes).
2. I’m going to address the mutator stuff in the blog post I write. Stay tuned…
3. My model is correct–it’s mutation RATE per nucleotide ~ thetaS, not mutations~thetaS. The graph is actually mutations~thetaS*gene length, as Inigo states is appropriate, which is why the uniform rate hypothesis doesn’t graph as a perfectly straight diagonal line (variations in gene length). These results hold for both my thetaS estimates, as well as Inigo’s estimates. The heterogeneous mutation rate hypothesis captures the expectation that genes with very high estimated thetaS should have more synonymous mutations in the LTEE than genes with very low estimated thetaS. There’s no evidence of this (if you don’t believe me, feel free to peruse my data and code and do whatever statistics you like!). I do think I treat both hypotheses even-handedly–I’m open to seeing alternative statistical treatments if you disagree, but I’d rather not redo the analysis as 1) I’m lazy and 2) I have more pressing stuff to work on.
Hope this makes things more clear!
Pingback: All the cool kids are on arXiv and Haldane’s Sieve .. why you should be too
See a full blog post by Rohan here — http://ivory.idyll.org/blog/rohan-on-weird-patterns.html
Pingback: Most viewed on Haldane’s Sieve: October 2012 | Haldane's Sieve
Hi there!
The paper above by Maddamsetti et al. was submitted to Nature as a comment to our paper and we were given the opportunity to respond. After examining our response, the editor decided not to publish this exchange. If you are interested, you can read our arguments in ArXiv: http://arxiv.org/abs/1211.0928
Thanks for reading!,
Inigo
Thanks Inigo. We’re posted the abstract of your response here:
I’m glad you posted your response to arXiv; this has been an instructive exchange.
Pingback: Most viewed on Haldane’s Sieve: November 2012 | Haldane's Sieve
Pingback: Haldane’s Sieve sifts through 2012 | Haldane's Sieve