
Genome Sequencing Highlights Genes Under Selection and the Dynamic Early History of Dogs
Adam H. Freedman, Rena M. Schweizer, Ilan Gronau, Eunjung Han, Diego Ortega-Del Vecchyo, Pedro M. Silva, Marco Galaverni, Zhenxin Fan, Peter Marx, Belen Lorente-Galdos, Holly Beale, Oscar Ramirez, Farhad Hormozdiari, Can Alkan, Carles Vilà, Kevin Squire, Eli Geffen, Josip Kusak, Adam R. Boyko, Heidi G. Parker, Clarence Lee, Vasisht Tadigotla, Adam Siepel, Carlos D. Bustamante, Timothy T. Harkins, Stanley F. Nelson, Elaine A. Ostrander, Tomas Marques-Bonet, Robert K. Wayne, John Novembre
(Submitted on 31 May 2013)
To identify genetic changes underlying dog domestication and reconstruct their early evolutionary history, we analyzed novel high-quality genome sequences of three gray wolves, one from each of three putative centers of dog domestication, two ancient dog lineages (Basenji and Dingo) and a golden jackal as an outgroup. We find dogs and wolves diverged through a dynamic process involving population bottlenecks in both lineages and post-divergence gene flow, which confounds previous inferences of dog origins. In dogs, the domestication bottleneck was severe involving a 17 to 49-fold reduction in population size, a much stronger bottleneck than estimated previously from less intensive sequencing efforts. A sharp bottleneck in wolves occurred soon after their divergence from dogs, implying that the pool of diversity from which dogs arose was far larger than represented by modern wolf populations. Conditional on mutation rate, we narrow the plausible range for the date of initial dog domestication to an interval from 11 to 16 thousand years ago. This period predates the rise of agriculture, implying that the earliest dogs arose alongside hunter-gathers rather than agriculturists. Regarding the geographic origin of dogs, we find that surprisingly, none of the extant wolf lineages from putative domestication centers are more closely related to dogs, and the sampled wolves instead form a sister monophyletic clade. This result, in combination with our finding of dog-wolf admixture during the process of domestication, suggests a re-evaluation of past hypotheses of dog origin is necessary. Finally, we also detect signatures of selection, including evidence for selection on genes implicated in morphology, metabolism, and neural development. Uniquely, we find support for selective sweeps at regulatory sites suggesting gene regulatory changes played a critical role in dog domestication.

The Supporting Information file is not currently available on arXiv. For now, you can access it via the following link: https://www.dropbox.com/s/2yoytspv1iods7s/Freedman_etal_SupportingInfo_arxiv.pdf
We’ve also made a few minor edits to the SI sections referred to in the manuscript. So, if you’ve downloaded the manuscript already, best to do so again to avoid confusion. Thanks, Adam Freedman
Really fun paper, glad to see it on the arXiv. A couple small comments:
1. Do you have an intuition about why the PSMC looks so different between dogs and wolves? Naively interpreting Figure 1D would seem to suggest that dogs and wolves had different about 800,000 years ago, but presumably they were the same species then.
about 800,000 years ago, but presumably they were the same species then.
2. It’s not obvious to me that the assumed phylogeny for parameter estimation (Figure S10.1) is consistent with the D-statistics (Table S9.4.1). Specifically, you get a fairly large, positive D statistic for D(Jackal, Boxer;Chinese wolf, Israeli wolf), indicating gene flow (possibly indirect gene flow) somewhere in that tree (btw either boxer and israeli wolf or btw jackal and chinese wolf); it’s not obvious how this happens in the phylogeny. You seem to have already done simulations to test this: in simulations from the optimized g-phocs model, are the D-statistics on the simulated data consistent with the observed ones?
Thanks Joe! Regarding your first question, we see something similar even in simulations (Supp Fig S9.2.3-9.2.5), albeit less pronounced. This suggests that these unexpected deep time Ne values may be a natural output of PSMC when it is applied to demographic models that include bottlenecks, though we are still exploring this in more detail, and would be curious if others have seen this behavior. It’s also worth noting the deep time Ne estimates are driven by the relative abundance of short segments with high heterozygosity. If some of these short segments are due to clusters of genotyping errors (specifically ones creating high rates of hets) and if the presence of these clusters vary across lineages, this could also contribute to the effect.
For the second question, I’m going to let Ilan Gronau weigh in here. He’s done great work on the demographic analyses in this paper, and helped lead us on synthesizing the G-PhoCS and the D-stats, so he’s very capable to weigh in here. Stay tuned!
Re. PSMC: In your simulations the deep time discrepancy looks like an edge effect, whereas it seems far more pronounced in the data, and if it wasn’t clearly at odds with what we know about dogs & wolves, I could imagine us assuming this was real. Something equally odd was in the recent polar bear paper (http://www.plosgenetics.org/article/info%3Adoi%2F10.1371%2Fjournal.pgen.1003345, Figure_S6), where curves from individuals within the same species diverge going back in time!
I think aspects of the output from PSMC is highly sensitive to data processing issues as well as demography (e.g. admixture which your Jackal data demonstrates very nicely), and a lot of care needs to be taken when interpreting them.
Very nice paper by the way!
Joe, in response to your question about consistency of the ABBA/BABA D-statistics with the gene flow model we infer using G-PhoCS, you can find a very brief discussion on this issue in Supp Section S10.3.1. We argue that the significant D-statistics we get for (Boxer – Israeli wolf) gene flow could be a result of gene flow from Basenji to Israeli wolf. Note that these significant D-statistics are obtained as long as Basenji is not used as the third non-outgroup sample. We confirm this hypothesis in the six separate analyses described in S10.3.1. When we assume a model with gene flow between Israeli wolf and all three dog populations, we infer significant gene flow only between Israeli wolf and Basenji (in both directions). The signal for (Boxer – Israeli wolf) gene flow is completely eliminated in this case, and only appears when we remove the migration band (Basenji–>Israeli wolf) from the model. We take this to imply that the significant D-statistics obtained for (Boxer – Israeli wolf) are more likely to be a consequence of gene flow from Basenji to Israeli wolf than a consequence of direct gene flow between Boxer and Israeli wolf.
We like your suggestion of using the simulated data to provide further support for our interpretation of the results. We will follow up on that. We actually gave considerable thought in integrating of the various aspects of the demographic analysis (PSMC, ABBA/BABA, and G-PhoCS) into a unified picture. We consider this to be an important feature of this study.
Hi John and Ilan,
Thanks for the responses! I’ll ask around about the PSMC behavior.
Regarding the consistency between the D statistics and the G-PhoCS model, I think showing that the simulated data recapitulate the observed tree violations would be stronger evidence that the model is consistent with the major aspects of history. Right now the evidence seems indirect, and in any case it’s a natural thing to look at since you’ve already run the simulations. I agree that integrating these different components of analysis are a novel aspect of the study; this seems like a promising direction.
I’ve been asked why the inferred split time between dogs and wolves in this paper (11-17kya) is so different than the estimate in the recent Wang et al. paper (32kya). I think this is almost entirely due to the assumed mutation rate calibration: Wang et al. assume a mutation rate of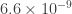 /generation, while Freedman et al. assume
/generation, while Freedman et al. assume 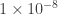 /generation. Converting both to the same mutation rate reduces the discrepancy considerably (if Wang et al. had used the Freedman mutation rate, they would instead get a split time of 21kya). Like in human demographic inferences, it seems a lot depends on getting this number pinned down!
/generation. Converting both to the same mutation rate reduces the discrepancy considerably (if Wang et al. had used the Freedman mutation rate, they would instead get a split time of 21kya). Like in human demographic inferences, it seems a lot depends on getting this number pinned down!
Totally agreed! In our discussion we tried to make clear the nontrivial affect uncertainty in mutation rate has on these dates. I’ll be excited to see more precise estimates from across a broader set of mammals and seeing pedigree-type rates estimated from dogs seems easily within reach. One additional point to add about our pre-print: Weiwei Zhai (a key author from the Wang et al paper) kindly pointed out to me that in the part of our discussion on the impact of assumed mutation rate we slipped up the units when we cite their assumed rate (using per year instead of per generation). It was a sentence that got added in a late stage draft and we didn’t catch the mistake in our last proofreading before posting. Having the extra eyes on this paper prior to publication has been great in this way – thanks Weiwei!
Thanks John, I’d missed that comment in the preprint. Agreed that direct estimates are well within reach now.
Pingback: The dog days of evolutionary genomics : Gene Expression
Pingback: Most viewed on Haldane’s Sieve: June 2013 | Haldane's Sieve
Pingback: Some preprint comment streams at Haldane’s sieve and related sites | Haldane's Sieve
Pingback: Sifting through 2013 with Haldane’s Sieve | Haldane's Sieve
Pingback: Author post: Genome Sequencing Highlights the Dynamic Early History of Dogs | Haldane's Sieve
Pingback: Most viewed on Haldane’s Sieve: January 2014 | Haldane's Sieve