This guest post is by Christopher Brown, Lara Mangravite, and Barbara Engelhardt on their paper: Integrative modeling of eQTLs and cis-regulatory elements suggest mechanisms underlying cell type specificity of eQTLs arXived here.
Why do we study eQTLs? Why don’t we count bristles?
The genetic dissection of complex trait models, independent of the particular phenotype, is useful for improving our understanding of the genetic architecture underlying the biochemical function that regulates complex traits in general. In the last ten years, gene expression levels themselves have emerged as useful phenotypes amenable to genetic dissection with several advantages, most notably that it is easy to accurately quantify tens of thousands of traits simultaneously (indeed even more when we address splicing and promoter usage). While the identification of SNPs that are associated with variation in gene expression (eQTLs) is certainly interesting at this basic level, an additional critical use for eQTL data has emerged. Because the majority of common human phenotypic variation appears to be driven by non-coding sequence variants, eQTL analyses are beginning to help with the mechanistic interpretation of GWAS results. In light of these interests and applications, we believe that eQTL analyses are hampered by (at least) three important limitations, which we have attempted to address in our recent preprint:
(1) Methodological (non) uniformity. Most eQTL studies have been performed by different groups, on different genotyping and gene expression platforms, with different association methods, and using different criteria for defining significance. This lack of uniformity complicates even simple cross study comparisons; for example, what fraction of genes has one or more independently associated eQTL when analyzed across tissues? We address this issue by testing for eQTL associations across a diverse set of cell types using a uniform pipeline with standardized analysis parameters to perform all analytical steps starting from raw data. As a fairly trivial example, our analyses across the eleven studies demonstrated that nearly all of the variation in the proportion of genes with significant eQTL associations identified within each study can be explained by just two factors: study size and replicate gene expression measurements. The proportion of genes with one or more independently associated eQTLs, then, is probably not 5-10% as has been hypothesized, but most or all of them, which we can get a better picture of when we design studies with sufficient power.
(2) Undercharacterized cell specificity. It is generally agreed upon that some eQTLs regulate gene expression in a cell type specific manner. When using eQTLs to interpret the genetic contribution to complex clinical traits, it is important to consider the cell type(s) most relevant to the trait of interest. However, if we don’t know what cell type is responsible for a phenotype or if we don’t have eQTL data for the cell type of interest, we are forced to extrapolate inferences about eQTLs derived from other cell types. By enabling the simultaneous comparison of within and between cell type eQTL replication for multiple cell type combinations and integrating these results with cis-regulatory element (CRE) mapping data from ENCODE, we have addressed several unresolved questions concerning the nature of cell type specific and ubiquitous eQTL SNPs. We find that eQTL-CRE overlap is frequently cell type specific and that this information can be used to predict cell specificity of eQTLs in the absence of additional gene expression data from the cell type of interest. While these results are certainly preliminary (and indeed we see many possible improvements), we hope this will improve the utility of eQTL-GWAS comparisons, particularly in situations where the GWAS cell type of interest lacks eQTL data.
(3) Resolution, causality, and mechanism. Lead tag SNPs are probably causal variants less than 30% of the time. While larger and more diverse genomic sample sets are essential to improve the resolution for identifying causal variants, this is not always possible due to time or budget constraints. However, the application of orthogonal genomic data also has the potential to considerably refine resolution with the added benefit of providing insight into the mechanism through which a causal variant acts. We approach this (as a few other groups have – notably Dan Gaffney et al.) by integrating CRE data into our analyses, because it appears that genetic variants that overlap certain types of CREs are much more likely to be functional than those that do not. We believe that this hypothesis, and the methods used to address it, need to be validated with directed functional assays, but we see no reason to doubt the principle of understanding heritable phenotypes using genotype functional analyses. Furthermore, the analysis of cell specific eQTL data in the context of cell specific CRE data, which is now possible, enables predictions about the regulatory mechanisms that are affected by a specific eQTL, which will allow us to place GWAS hits into pathways or provide other meaningful biological insights.
Why did we submit the paper to arXiv and Haldane’s Sieve?
We are big proponents of open access publication, open data, and transparent methods and analysis. At least part of what we’ve done here is to create a resource that we hope will be useful to the broader community. We are open to pre and post publication review of and commentary on our motivations and methods. Furthermore, we have submitted all of the eQTLs we identify to a database of eQTLs (eqtl.uchicago.edu), and we are currently securing funding to develop open access, online tools to help GWAS researchers follow up specific functional variants using our methods.
Christopher Brown, Lara Mangravite, Barbara Engelhardt

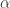 -proteobacterium Wolbachia and an unnamed
-proteobacterium Wolbachia and an unnamed 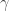 -proteobacterium from the Enterobacteriaceae. Microbial communities varied among host populations for measures of community diversity and exhibited significant population structure. We also uncovered a strong negative correlation in the abundance of the two dominant OTUs, suggesting they may fulfill similar roles as nutritional mutualists. This broad survey represents the most comprehensive assessment, to date, of the microbes that associate with bed bugs, and uncovers evidence for potential antagonism between the two dominant members of the bed bug microbiome.
-proteobacterium from the Enterobacteriaceae. Microbial communities varied among host populations for measures of community diversity and exhibited significant population structure. We also uncovered a strong negative correlation in the abundance of the two dominant OTUs, suggesting they may fulfill similar roles as nutritional mutualists. This broad survey represents the most comprehensive assessment, to date, of the microbes that associate with bed bugs, and uncovers evidence for potential antagonism between the two dominant members of the bed bug microbiome.