This guest post is by Anand Bhaskar and Yun Song on their paper: “The identifiability of piecewise demographic models from the sample frequency spectrum”. arXived here.
With the advent of high-throughput sequencing technologies, it has been of great interest to use genomic data to understand human demographic history. For example, we now estimate that modern humans migrated out of Africa around 60K-120K years ago [1,2], and that Neandertals may have admixed with modern humans in Europe as recently as 47,000 years ago [3]. Apart from satisfying curiosity about our anthropological history, the inference of demography is important for several scientific reasons. Most importantly, demographic processes influence genetic variation, and understanding the interplay between natural selection, genetic drift, and demography is a key question in population genetics. Also, controlling for demography is important for practical applications. For example, the demography inferred from neutrally evolving genomic regions can serve as a null model when searching for regions under selection. Demographic models could also be used to circumvent the problem of spurious associations in case-control studies induced by population substructure.
A summary of whole haplotypes that is commonly used in population genetic analyses is the sample frequency spectrum (SFS). For a sample of n haplotypes from a panmictic (i.e. without substructure) population, the SFS is an (n-1)-dimensional vector where the i-th entry is the proportion of SNPs with i copies of the mutant allele in the sample. One can talk about a mutant/derived allele because most analyses assume that mutations are rare enough that the observed SNPs are dimorphic. The first few entries of the SFS capture the proportion of rare SNPs in the sample and are especially useful for inferring recent population history. Several recent large sample sequencing studies [4-6] have found that humans have many more putatively neutral rare SNPs compared to predictions from a constant population size model. Using the SFS from their data, these studies all infer demographic models with recent exponential population expansion.
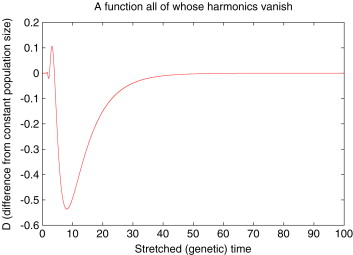
However, until fairly recently, it was not known whether the SFS of a sample uniquely determines the underlying demographic model. Could it be possible that two different demographic models produce the exact same expected SFS for all sample sizes? In 2008, Simon Myers, Charles Fefferman, and Nick Patterson came up with an elegant mathematical argument [7] to show that there are infinitely many population size histories that generate the same expected SFS for all sample sizes. They even provided an explicit example of a population size history which produced the same expected SFS as a constant population size model. However, their example history had increasingly rapid oscillations in the population size in the recent past, something that we might not expect to find in real biological populations. After all, even though we commonly use continuous-time models of evolution like coalescent theory and diffusion processes, biological populations evolve in discrete events of birth and death.
Our research group has been working on demographic inference from the SFS and from full sequence data for the last several years, and so it was natural for us to ask whether the class of population size histories that are commonly inferred using statistical algorithms might also suffer from this non-identifiability problem. Most statistical methods infer piecewise population size histories, where the pieces come from some biologically-motivated family of functions. In particular, piecewise constant and piecewise exponential models commonly appear in the literature. And if one can indeed uniquely identify piecewise demographic models from the SFS, what sample sizes are needed to do so?
In our paper, we address this question by proving that if the underlying population size function is piecewise with at most K pieces, then the expected SFS of a random sample of size n uniquely determines the demography as long as n is larger than some function of K that depends on the type of pieces of the population size function. For example, if the underlying demographic model was piecewise constant with at most K pieces (i.e. described by at most 2K – 1 parameters), then the expected SFS of a sample of size 2K uniquely determines the demographic model. In other words, no two piecewise constant population size functions with at most K pieces can generate the same expected SFS for a sample of size 2K or larger. For piecewise exponential demographic models with at most K pieces, a sample size of 4K – 1 is sufficient to uniquely determine the demographic model. When one doesn’t know which allele is ancestral and which is derived (for example, if outgroup information is lacking at the relevant SNPs), demographic analysis can still be carried out using the SFS by “folding” it. The folded SFS has floor(n/2) entries, where the i-th entry is the proportion of SNPs with i copies of the minor allele (which might be an ancestral or derived allele). Since the folded SFS has only roughly half the dimension as the full SFS, one might expect to require twice as many samples to uniquely determine the demographic model from the folded SFS compared to the full SFS. We formally prove in our paper that this intuition is indeed correct.
It is important to stress that this identifiability result is statistical rather than algorithmic in that that one would need to have perfect information about the expected SFS of a random sample in order to uniquely determine the underlying piecewise demography. In practice, one can get good estimates of the expected SFS by considering a large number of SNPs in the inference procedure, and by considering SNPs that are farther apart along the chromosomes so that the coalescent trees for the sample at different SNPs will be roughly independent of each other. More work is certainly needed to understand how much genomic data (measured both in terms of the number of SNPs and the sample size) would be needed in practice to robustly infer realistic demographic models.
Works cited:
[1] Li, H. and Durbin, R. (2011) Inference of human population history from individual whole-genome sequences. Nature 475, 493–496.
[2] Scally, A. and Durbin, R. (2012). Revising the human mutation rate: implications for understanding human evolution. Nature Reviews Genetics, 13(10), 745-753.
[3] Sankararaman, S., Patterson, N., Li, H., Pääbo, S., and Reich, D. (2012) The date of interbreeding between Neandertals and modern humans. PLoS Genetics 8, e1002947.
[4] Nelson, Matthew R., et al. (2012) An abundance of rare functional variants in 202 drug target genes sequenced in 14,002 people. Science 337, 100–104.
[5] Tennessen, Jacob A., et al. (2012) Evolution and functional impact of rare coding variation from deep sequencing of human exomes. Science 337, 64–69.
[6] Fu, Wenqing, et al. (2012) Analysis of 6,515 exomes reveals the recent origin of most human protein-coding variants. Nature 493, 216–220.
[7] Myers, S., Fefferman, C., and Patterson, N. (2008) Can one learn history from the allelic spectrum? Theoretical Population Biology 73, 342–348.