This guest post is by Frank Albert and Leonid Kruglyak on their preprint (with co-authors) “Genetic influences on translation in yeast“.
This post is on a manuscript we recently posted to both biorXiv and arXiv on how genetic differences between yeast strains influence protein translation. Here, we give some background for the project and what the results mean in our eyes. We also highlight a few interesting aspects of regression analysis that may be useful to other researchers in genomics, but aren’t currently widely appreciated (at least they weren’t obvious to us before we dove into these data). We appreciate any comments and thoughts!
Protein translation and the genetics of gene expression
Our study was motivated by earlier reports that regulatory variation among individuals in a species can have surprisingly different effects on mRNA vs. on protein levels. For those not fully up to speed on these questions, here’s a quick recap. Individuals differ from each other in many aspects (e.g. in appearance and disease susceptibility), and for many traits, these differences are at least in part due to genetic variation. Research in genetics (both in humans and in other species) is focused on identifying DNA sequence variants in the genome that contribute to phenotypic variation, and on understanding the molecular mechanisms through which these variants alter traits. One general class of such mechanisms is alteration of gene expression, and genetic loci that affect gene expression are known as “expression quantitative trait loci” (eQTL). DNA sequence variants can change expression levels of genes in a number of ways, and some of the expression differences in turn are thought to alter organismal traits. Large and growing eQTL catalogues exist for several species. However, to measure “gene expression”, virtually all these studies measure mRNA rather than protein levels. This isn’t because mRNA is the more relevant molecule to look at, but primarily because mRNA is much easier and cheaper to measure on a global basis than are proteins. There have been a few studies in model organisms and—more recently— in humans that examined genetic variants that influence protein levels. Their results were surprising: the loci that influenced proteins (protein QTL, pQTL) were apparently often different from those that influenced mRNA levels, and vice versa. These results were both troubling and exciting. They were troubling because if genetic changes in protein levels are not a faithful reflection of effects on mRNA levels, the relevance of mRNA-based eQTL maps is less obvious. The results are exciting from a basic science perspective, because they suggest that lots of genetic variants that specifically influence posttranscriptional processes remain to be discovered.
Our current paper begins to explore these issues by measuring one such posttranscriptional process: protein translation, the process by which mRNA molecules are read by ribosomes and “translated” into peptide chains. Translation is interesting because there is literature that suggests that its regulation is a major determinant of protein levels, perhaps as important as the regulation of mRNA levels. Translation is also convenient because it is the last step along the gene expression cascade that can still be assayed using the high throughput sequencing technologies that underlie much of the current boom in genomics in general and eQTL detection in particular. The trick is to isolate only those bits of mRNA that sit inside of ribosomes as they march along the mRNAs during translation. These “ribosome footprints” can then be sequenced, counted, and compared to mRNA levels measured in parallel to get a quantitative readout of how much each gene is being translated.
For our current paper, we teamed up with Jonathan Weissman at UCSF. The Weissman lab pioneered measuring translation by sequencing, and Dale Muzzey (a postdoc with Jonathan) generated the data for our current experiment. We chose a very simple but powerful design: we compared translation in two strains of yeast that are genetically different from each other, and also measured allele-specific translation in the diploid hybrid between these two strains. Using the strain comparison, we can quantify the aggregate effect of all genetic differences between the two strains on translation. In the hybrid, we can specifically see the effects of those variants that act in cis, an important sub-group of eQTL.
We encourage you to read the detailed results in the paper, but in a nutshell, we found that genetic differences clearly do have an effect on translation—but not a terribly large one. Genes that differ in mRNA abundance typically also differ in footprint abundance, and the effect of translation was typically to subtly modulate mRNA differences, rather than erase them or create protein differences from scratch. While there are some exceptions, the take-home message is that on average, differences in protein synthesis are reasonably well approximated by differences in mRNA levels.
Thus, translation does not appear to create major discrepancies between eQTL and pQTL. So what explains such reported discrepancies? Without going into great detail, we think that at least a part of the explanation is that those discrepancies may have been overestimated. This fits with our recent paper in which we used extremely large yeast populations to map pQTL with higher statistical power, and recovered many pQTL at sites where earlier work had found an eQTL, but no pQTL. Are pQTL in most cases simply a reflection of eQTL? It is too early to tell, and we’ll need improved designs and datasets to answer to answer this question with certainty. At the very least, our recent work suggests that genetic influences on protein levels more accurately reflect those on mRNA levels than previous reports had suggested.
“Spurious” correlations and adventures in line fitting
While we worked on the translation paper, we encountered a few technical aspects that are worth sharing in some detail. Specifically, a natural question to ask is how differences in translation compare to differences in mRNA levels. When a gene differs in mRNA abundance between strains, does translation typically lead to a stronger or weaker footprint difference? Does translation typically “reinforce” or “buffer” mRNA differences (or is there no preference either way)? An intuitively attractive analysis is to plot the mRNA differences versus differences in “translation efficiency”, or TE (i.e. the amount of ribosome footprints divided by the amount of mRNA for a given gene). This comparison is shown in Figure 1 for our hybrid data.

In the figure, differences are plotted as log2-transformed fold changes so that zero indicates “no change”, and the resulting distributions of differences are more or less normally distributed. We see a seductively strong negative correlation between mRNA differences and TE differences. Taken at face value, this seems to suggest that mRNA differences are typically accompanied by a difference in TE that buffers the mRNA difference: more mRNA in one strain is counteracted by lower TE, presumably resulting in a protein difference that is smaller than the mRNA difference would predict.
However, this analysis is misleading. The key point is that the TE difference is not independent from the mRNA difference. In log2 space, the TE difference is simply the footprint difference minus the mRNA difference (in non-log space, the TE difference is the footprint difference divided by the mRNA difference). Therefore, the larger the mRNA difference becomes, the smaller the TE difference becomes simply by definition. The fact that the correlation between TE differences and mRNA differences is negative is by itself not informative about the relationship between differences in translation and mRNA levels. This can further be illustrated in Figure 2 (which is Supplementary Figure S4 in our preprint). Here, we simply draw two uncorrelated samples a and b from a normal distribution. The plot of b – a over a has a strong negative correlation, although there is no systematic relationship at all between these two quantities.

It turns out that the problems with comparing ratios (such as the TE difference) to their components (e.g., the mRNA difference, which serves as the denominator in computing TE) have been noted a long time ago (e.g., Karl Pearson termed them “spurious” correlations in 1897). They are worth remembering when carrying out functional genomic and systems biology data analyses in which multiple classes of molecules (mRNA, proteins, metabolites, etc.) are compared to each other and to ratios between them.
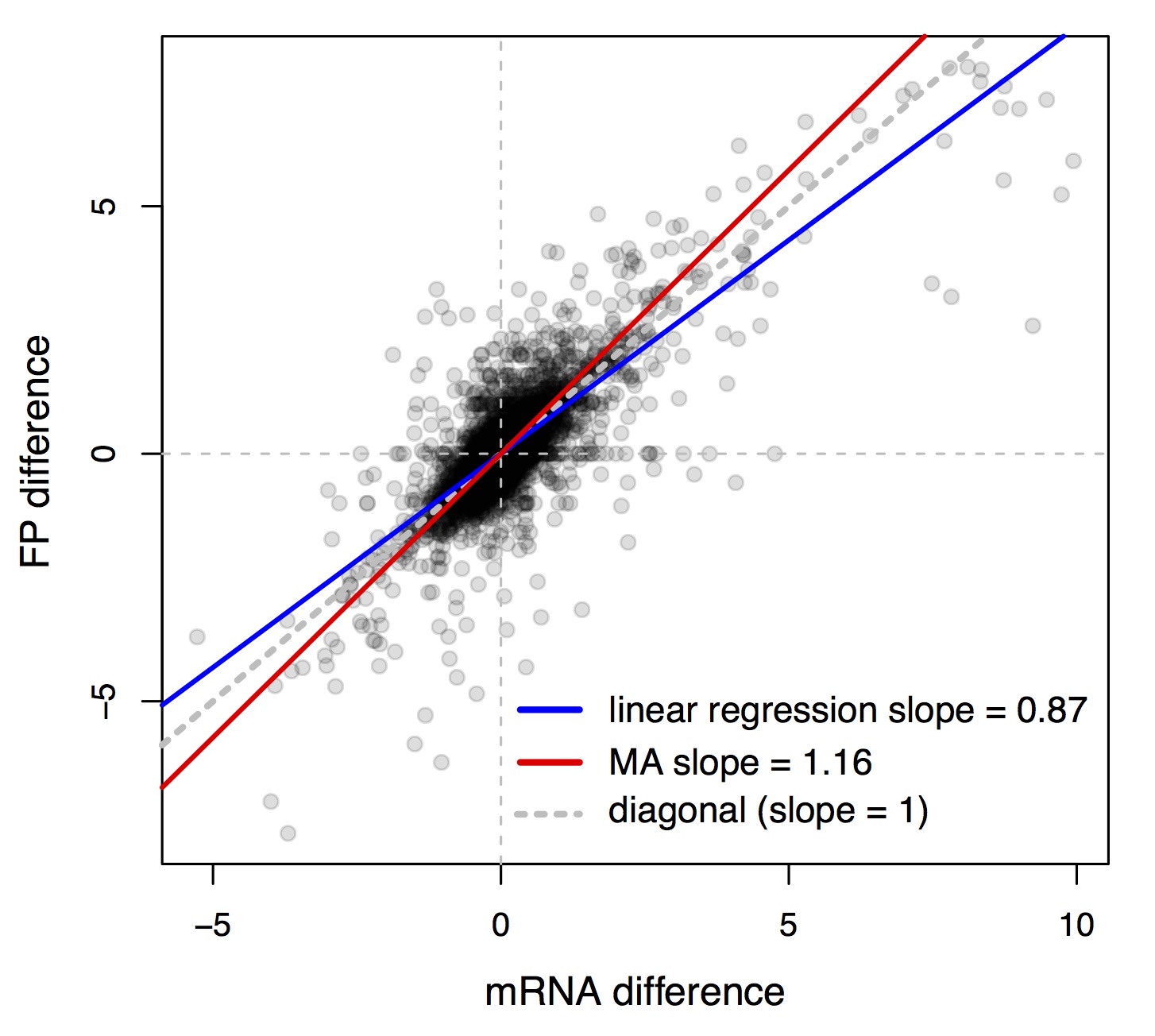
Because of this effect, we directly analyzed differences in the two quantities we measured: mRNA and ribosome footprint abundance. The slope of a linear regression between these two quantities was less than one (the blue line in Figure 3, which shows the data from the strain comparison). This might once again suggest a predominance of buffering interactions—for any given mRNA difference, we would predict a smaller footprint difference.

However, this inference is again misleading, because regression to the mean ensures that even if we measure the same quantity twice with some measurement noise (which is usually unavoidable), the slope of the regression line between these two replicates is always less than one. This is because when the first measure for a given gene is by chance larger than its true value, the measure is likely to be smaller in the second replicate. We estimated the regression slope between footprint differences and mRNA differences that would be expected if there were no preference for reinforcing or buffering interactions by using a randomization test that you can read about in the manuscript. The upshot is that the observed regression slope was steeper than those in the randomized data. We therefore concluded that on average, translation more often reinforces than buffers mRNA differences.
A related issue (pointed out to us by J.J. Emerson at UC Irvine) is that linear regression fits a line by minimizing only the error on the y-axis. This makes linear regression ideal for predicting y from x, but less than ideal for measuring the relationship (the slope) between x and y. For our purposes, it is better to use a different way of fitting lines to bivariate data: “Major Axis Estimation”. You can read all about MA in this great review, but briefly, the idea is to fit a line that minimizes the perpendicular distance of points from the line. The minimized error gets distributed between the y and the x axis, resulting in fitted lines that, unlike regression, provide unbiased estimates of the slope, and hence of the true linear relationship between x and y. Using MA on our data did not alter the conclusions we arrived at by using regression together with the randomization test, but it did provide more directly interpretable values for the various slopes. For example, the slopes in the randomized datasets were now centered on 1, whereas they had been degraded to be less than one using linear regression. The MA slope in our data was greater than one (the red line in Figure 3), supporting the inference of reinforcement.
These points on “spurious” correlations, regression to the mean and MA will be obvious to some (they were not to us going in), but we suspect and hope that they may prove useful for others working on genomic datasets.