This author post is by Cyrus Maher and Ryan Hernandez on their preprint A MOSAIC of methods: Improving ortholog detection through integration of algorithmic diversity, arXived here.
Rigorous evolutionary analysis of protein coding regions often requires high-quality multiple sequence alignments. These alignments can only be generated after the identification of orthologous sequences. In our pre-print, “A MOSAIC of methods: Improving ortholog detection through integration of algorithmic diversity”, we present a novel method that substantially improves the number and quality of detected orthologs, especially in the presence of sequencing error and complex evolutionary processes.
This endeavor grew out of our forthcoming work on the evolutionary impact of ancient pathogens on the human genome. Early on, we observed the decisive influence ortholog quality exerted on our downstream conclusions. As one might imagine, accurate sequence analysis is a fool’s errand if the sequences are, in fact, the wrong ones! Such experiences have impelled us to take a keen interest in orthologs, much as a bad case of gastroenteritis might inspire a sushi chef to become thoroughly attentive to the quality of his or her fish.
Identifying orthologous sequences is referred to as ortholog detection (OD). In brief, existing OD methods can be classified as tree-based, graph-based, or a hybrid of the two. Tree-based methods may use reconciliation techniques between gene and species trees or may rely on the gene tree alone. Graph-based methods can employ a variety of metrics to quantify similarity between sequences. Popular measures include sequence identity and matrix-weighted similarity scores. Syntenic information may also be incorporated in this context.
Here we consider alignments from UCSC (MZ), MultiParanoid (MP), translated BLAT (BL), and OMA. To briefly summarize the strengths of the considered methods: MZ utilizes syntenic similarity, MP includes all-by-all similarity in its calculations, OMA considers phylogenetic information directly, and BL does not require an accurately predicted proteome. In figure 1A of our paper, we illustrate the head-to-head performance of four popular methods for OD. Interestingly, we find striking complementarity between methods, motivating a search for a practical way to integrate ortholog predictions from methodologically diverse sources.

Figure 1: Comparison of sequence identity levels between methods A.) Heat map of the percent of orthologs for which BLAT (BL), OMA (OMA), MultiParanoid (MP),, and MultiZ (MZ) outperform one another. Performance is based on percent identity of each method’s orthologs to the human sequence. One method is considered to outperform another method if it improves percent identity by at least five percentage points. Text in diagonal cells shows the number of orthologs identified by each method, colored by the percent of transcripts at which a given method outperforms all the others
These efforts culminate in the presentation of MOSAIC, or Multiple Orthologous Sequence Analysis and Integration by Cluster optimization. MOSAIC is a well-documented python package that can flexibly integrate ortholog predictions from an arbitrary number of sources. We compare integrated MOSAIC alignments to those generated using each constituent method alone. Relative to the best-performing single method, we show that MOSAIC more than quintuples the number of sequences for which all orthologs of interest are successfully identified (see figure below). However, this increase in putative orthologs could be the result of, e.g. the improper inclusion low-quality or paralogous sequences. This does not appear to be the case for MOSAIC. Crucially, improvements in power are secured while simultaneously maintaining or improving functional-, phylogenetic-, and sequence identity-based measures of ortholog quality.
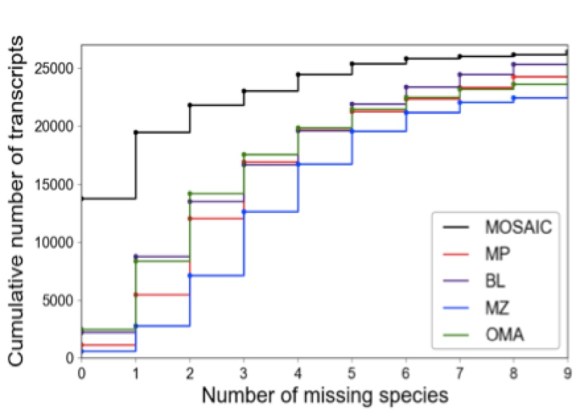
Figure 2: OD power and the effect of pooling methods A.) The cumulative number of human transcripts as a function of the maximum number of missing species allowed
These results are obtained from alignments between the human proteome and orthologs from nine species encompassing a range of primates and closely related mammals. For other sequence sets, the best strategy for method integration may differ slightly depending on, e.g. the level of divergence between species of interest. To account for this, MOSAIC provides several options for scoring and optimization, and even facilitates the specification of user-defined metrics for sequence similarity and cluster optimality.
In the future, we would also like to add functionality to automatically fetch relevant alignments from major ortholog databases. In the meantime, we hope that this tool will prove a useful addition to a variety of evolutionary analysis pipelines. We of course welcome feedback on how we might improve the performance and practical utility of the method. Thank you in advance for your input!