This post is by M.Mar Albà on her preprint (with co-authors) available from arRxiv Long non-coding RNAs as a source of new peptides.
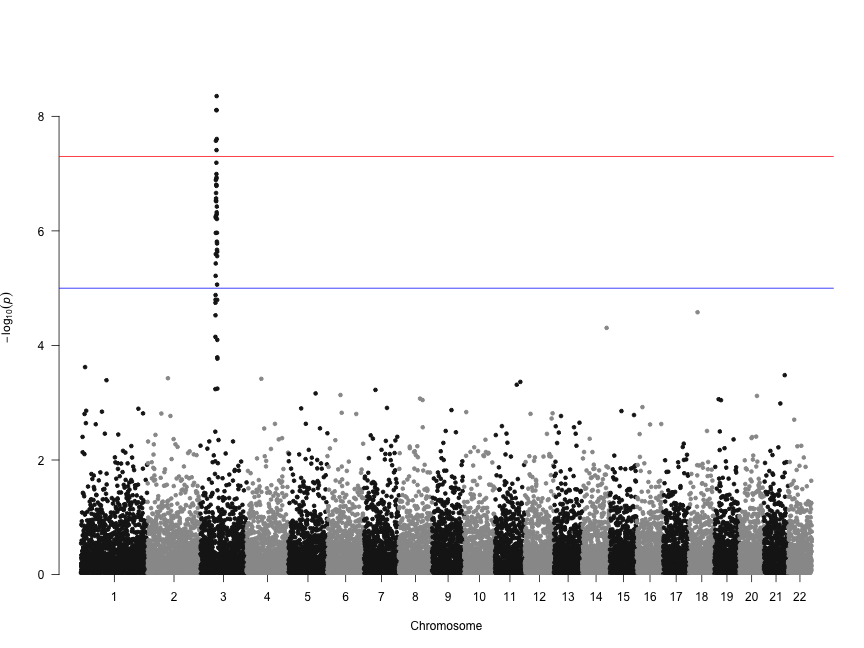
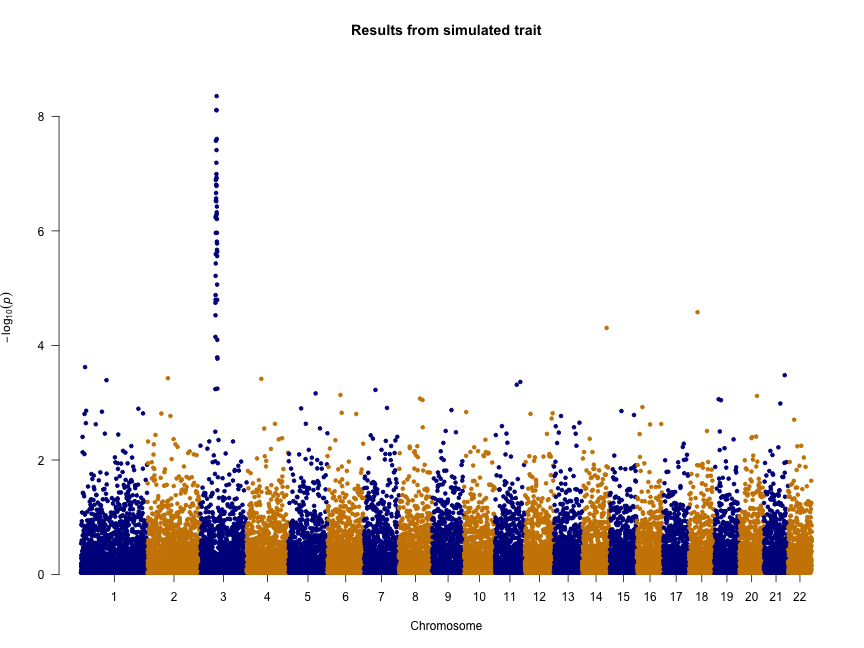
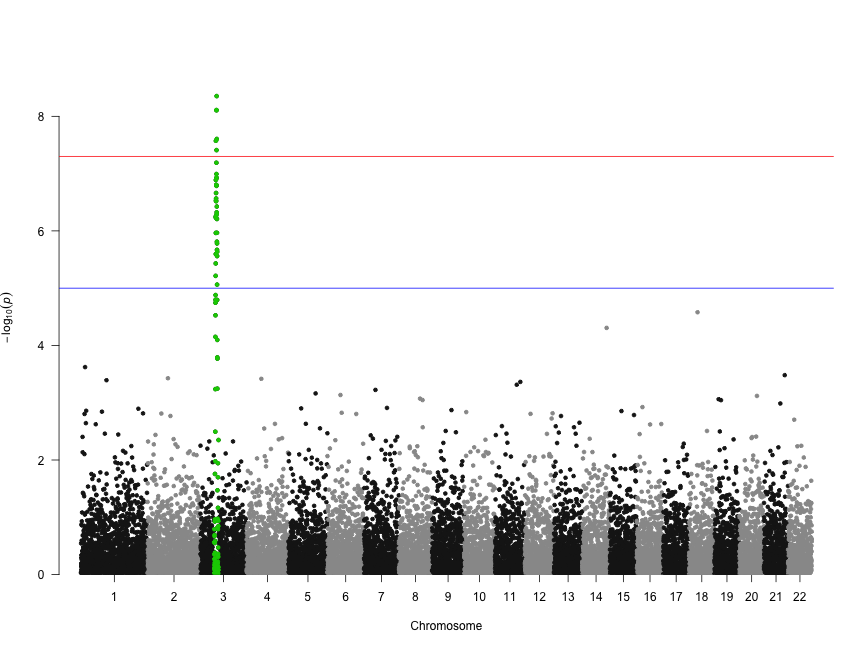
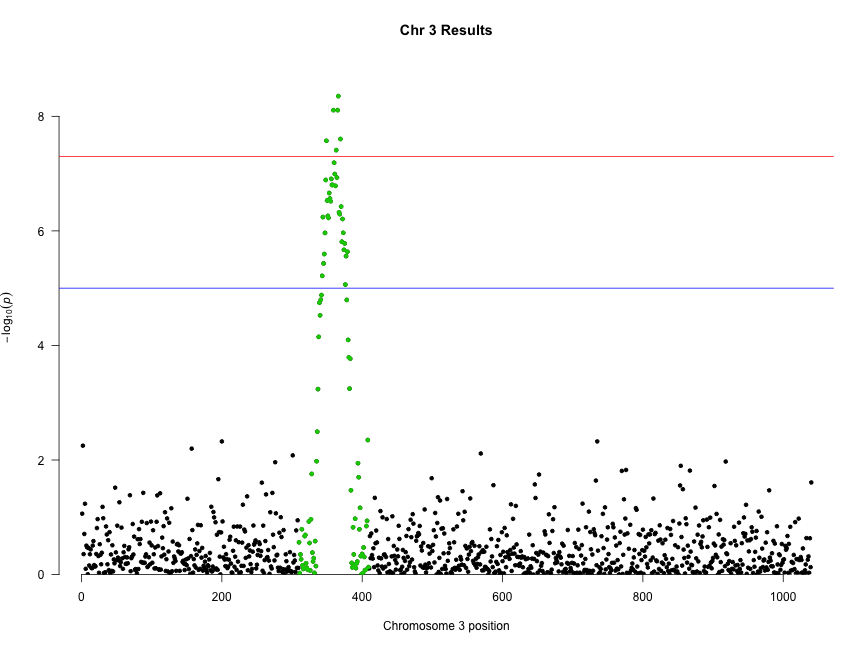
Several recent studies based on deep sequencing of ribosome protected fragments have reported that many long non-coding RNAs (lncRNAs) associate with ribosomes (see for example Everything old is new again: (linc)RNAs make proteins! a comment by Stephen M Cohen). We have analyzed the original data from experiments performed in six different eukaryotic species and confirmed that this is a widespread phenomenon. This is paradoxical because lncRNAs apparently have very little coding capacity with only short open reading frames (ORFs) that do not show sequence similarity to known proteins.
In contrast to typical mRNAs, many lncRNAs are lineage-specific. Therefore, if they are translated, they should be similar to recently evolved protein-coding genes. This is exactly what we have found. It turns out that transcripts encoding young proteins show very similar properties to lncRNAs; short and non-conserved ORFs, low coding sequence potential, and relatively weak selective constraints.
Evidence has accumulated in recent years that new protein-coding genes are continuously evolving (The continuing evolution of genes by Carl Zimmer). The birth of a new functional protein is a process of trial and error that most likely requires the expression of many transcripts that will not survive the test of time. LncRNAs seem to fit the bill for this role.