Long non-coding RNAs as a source of new peptides
Jorge Ruiz-Orera, Xavier Messeguer, Juan A. Subirana, M.Mar Albà
(Submitted on 16 May 2014)
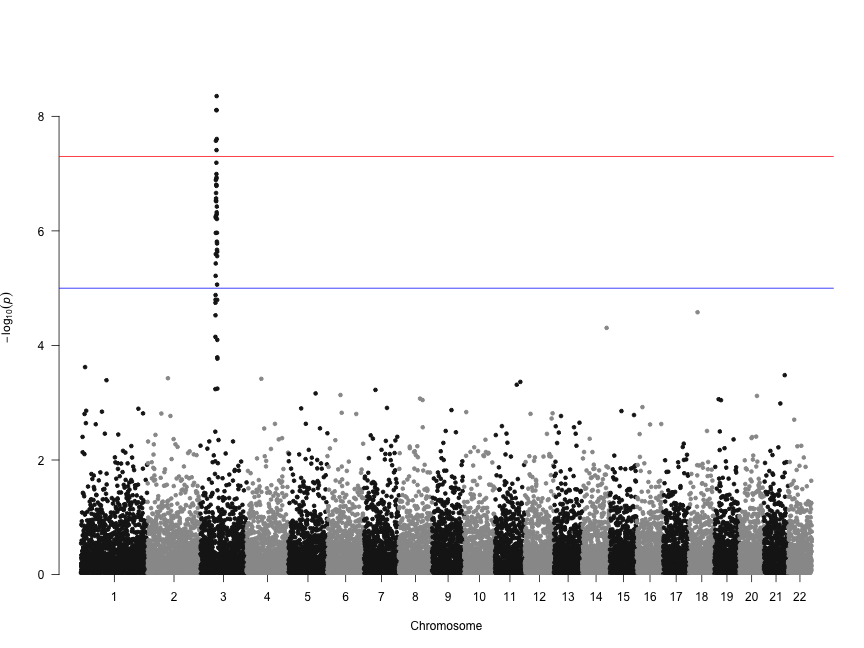
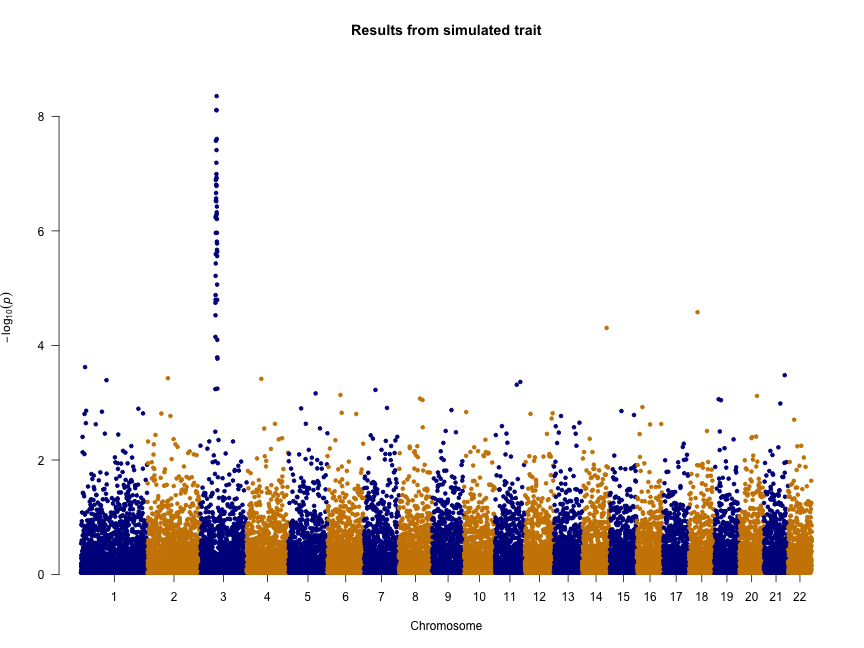
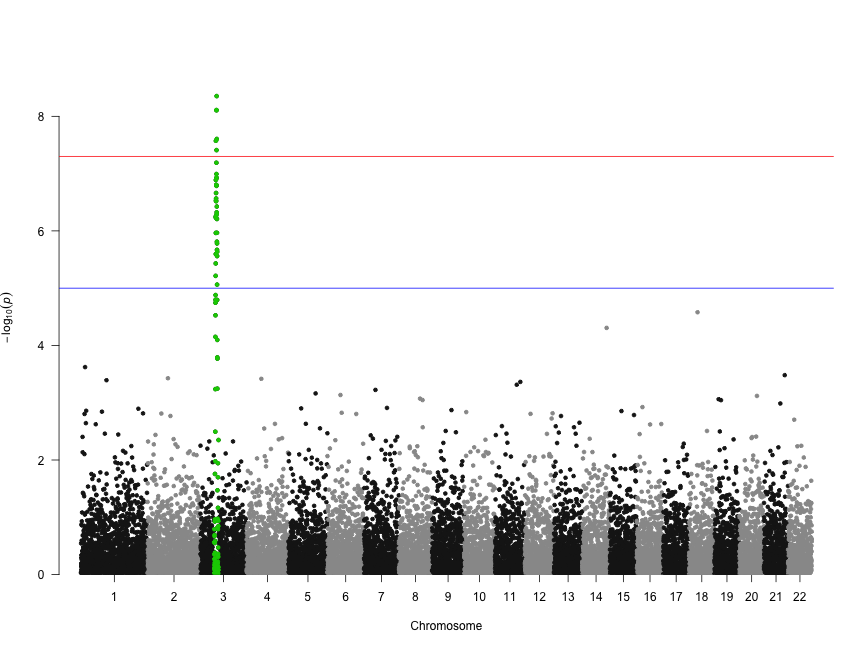
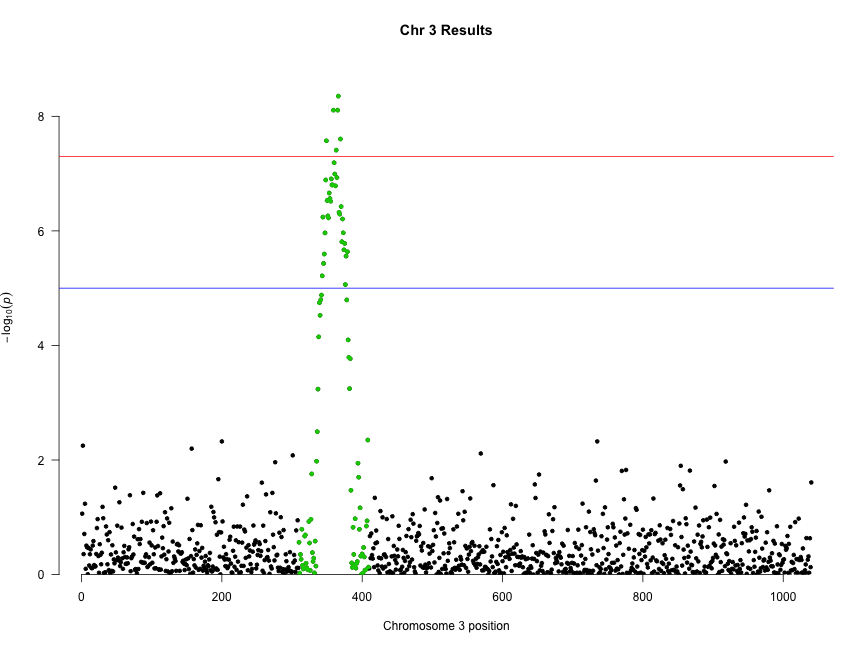
Deep transcriptome sequencing has revealed the existence of many transcripts that lack long or conserved open reading frames and which have been termed long non-coding RNAs (lncRNAs). Despite the existence of several well-characterized lncRNAs that play roles in the regulation of gene expression, the vast majority of them do not yet have a known function. Motivated by the existence of ribosome profiling data for several species, we have tested the hypothesis that they may act as a repository for the synthesis of new peptides using data from human, mouse, zebrafish, fruit fly, Arabidopsis and yeast. The ribosome protection patterns are consistent with the presence of translated open reading frames (ORFs) in a very large number of lncRNAs. Most of the ribosome-protected ORFs are shorter than 100 amino acids and usually cover less than half the transcript. Ribosome density in these ORFs is high and contrasts sharply with the 3UTR region, in which very often there is no detectable ribosome binding, similar to bona fide protein-coding genes. The coding potential of ribosome-protected ORFs, measured using hexamer frequencies, is significantly higher than that of randomly selected intronic ORFs and similar to that of evolutionary young coding sequences. Selective constraints in ribosome-protected ORFs from lncRNAs are lower than in typical protein-coding genes but again similar to young proteins. These results strongly suggest that lncRNAs play an important role in de novo protein evolution.