This guest post is by David Castellano and Adam Eyre-Walker on their preprint (with co-authors) Adaptive evolution is substantially impeded by Hill-Robertson interference in Drosophila.
Our paper “Adaptive evolution is substantially impeded by Hill-Robertson interference in Drosophila”, in which we investigate the role of both the rate of recombination and the mutation rate on the rate of adaptive amino acid substitutions, has been available at biorxiv (http://dx.doi.org/10.1101/021600) since 27 June.
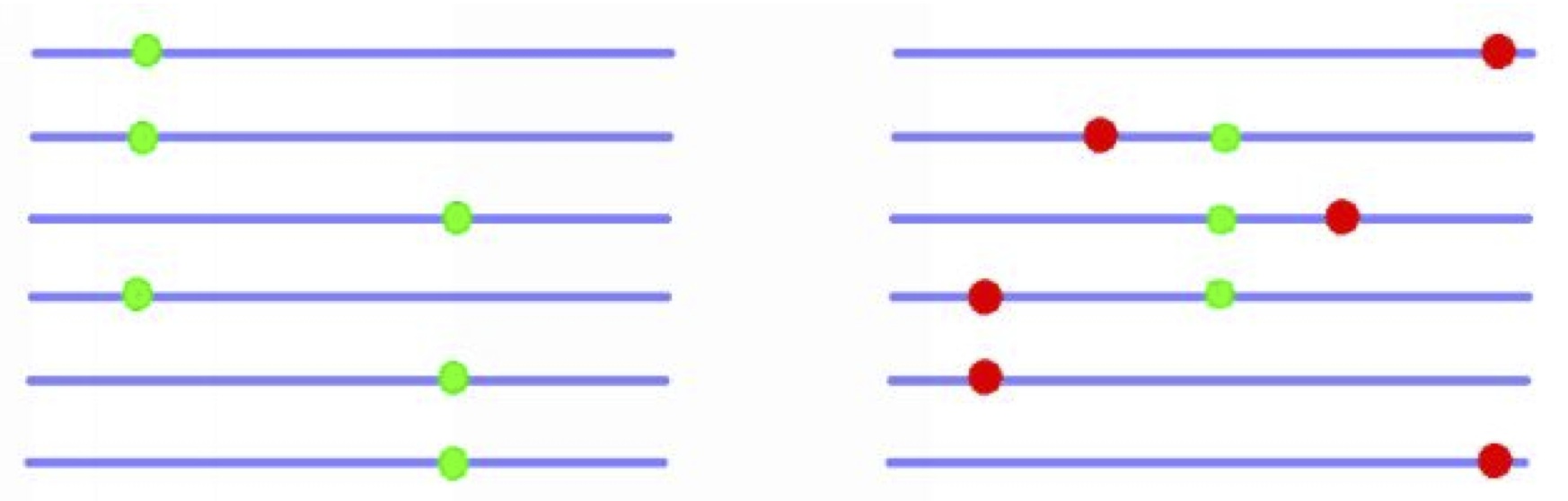
Population genetics theory predicts that the rate of adaptive evolution should depend upon the rate of recombination; genes with low rates of recombination will suffer from Hill-Robertson interference (HRi) in which selected mutations interfere with each other (see the figure below): a newly arising advantageous mutation may find itself in competition for fixation with another advantageous mutation at a linked locus on another chromosome in the population, or in linkage disequilibrium with deleterious mutations, which will reduce its probability of fixation if it can not recombine away from them.

A schematic HRi example among adaptive alleles (left) and among adaptive and deleterious alleles (right).
Likewise, it is expected that genes with higher mutation rates will undergo more adaptive evolution than genes with low mutation rates. More interestingly an interaction between the rate of recombination and the rate of mutation is also expected; HRi should be more prevalent in genes with high mutation rates and low rates of recombination. No attempt has been done so far to quantify the overall impact of HRi on the rate of adaptive evolution for any given genome. In our paper we propose a way to quantify the number of adaptive substitutions lost due to HRi – approximately 27% of all adaptive mutations, which would go to fixation since the split of D. melanogaster – D. yakuba if there was free recombination, are lost due to HRi. Moreover, we are able to estimate how the fraction of lost adaptive amino acid substitutions to HRi depends on gene’s mutation rate. In agreement with our expectations, genes with high mutation rates lose a significantly higher proportion of adaptive substitutions than genes with low mutation rates (43% vs 11%, respectively).
An open question is to what extent HRi affects rates of adaptive evolution in other species. Moreover, the loss of adaptive substitutions to HRi can potentially tell us something important about the strength of selection acting on some advantageous mutations, since weakly selected mutations are those that are most likely to be affected by HRi. This will require further analysis and population genetic modeling, but in combination with other sources of information, for example, the dip in diversity around non-synonymous substitutions, the site frequency spectrum the high frequency variants that are left by selective sweeps it may be possible to infer much more about the DFE of advantageous mutations than previously thought.
It will be of great interest to do similar analyses to those performed here in other species.
Comments very welcome!
David and Adam