
Genome-wide targets of selection: female response to experimental removal of sexual selection in Drosophila melanogaster
Paolo Innocenti, Ilona Flis, Edward H Morrow
Despite the common assumption that promiscuity should in general be favored in males, but not in females, to date there is no consensus on the general impact of multiple mating on female fitness. Notably, very little is known about the genetic and physiological features underlying the female response to sexual selection pressures. By combining an experimental evolution approach with genomic techniques, we investigated the effects of single and multiple matings on female fecundity and gene expression. We experimentally manipulated the mating system in replicate populations of Drosophila melanogaster by removing sexual selection, with the aim of testing differences in short term post-mating effects of females evolved under different mating strategies. We show that monogamous females suffer decreased fecundity, a decrease that was partially recovered by experimentally reversing the selection pressure back to the ancestral promiscuous state. The post-mating gene expression profiles of monogamous females differ significantly from promiscuous females, involving 9% of the genes tested. These transcripts are active in several tissues, mainly ovaries, neural tissues and midgut, and are involved in metabolic processes, reproduction and signaling pathways. Our results demonstrate how the female post-mating response can evolve under different mating systems, and provide novel insights into the genes targeted by sexual selection in females, by identifying a list of candidate genes responsible for the decrease in female fecundity in the absence of promiscuity.
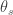
 ) across 2,930 orthologous gene alignments from 34 Escherichia coli genomes, and found substantial variation among genes in the density of synonymous polymorphisms. They argue that this pattern reflects variation in the mutation rate per nucleotide (
) across 2,930 orthologous gene alignments from 34 Escherichia coli genomes, and found substantial variation among genes in the density of synonymous polymorphisms. They argue that this pattern reflects variation in the mutation rate per nucleotide (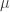 ) among genes. However, the effective population size (N) is not necessarily constant across the genome. In particular, different genes may have different histories of horizontal gene transfer (HGT), whereas Martincorena et al. used a model with random recombination to calculate
) among genes. However, the effective population size (N) is not necessarily constant across the genome. In particular, different genes may have different histories of horizontal gene transfer (HGT), whereas Martincorena et al. used a model with random recombination to calculate  . They did filter alignments in an effort to minimize the effects of HGT, but we doubt that any procedure can completely eliminate HGT among closely related genomes, such as E. coli living in the complex gut community.
. They did filter alignments in an effort to minimize the effects of HGT, but we doubt that any procedure can completely eliminate HGT among closely related genomes, such as E. coli living in the complex gut community.