For next guest post Fabian Staubach and Dmitri Petrov write about their paper (along with coauthors) Bacterial diversity associated with Drosophila in the laboratory and in the natural environment arXived here.
Host associated bacterial communities are ubiquitous, have a variety of effects on the host phenotype and play a role in host adaptation to new environments. Some clear examples of such adaptations are known but generally these are ancient associations between host and symbiont, such as the association between aphids and the obligate symbiotic bacterium Buchnera that provides the aphid with essential amino acids or the association between bee wolfs and Streptomyces that protects bee wolf larvae from fungal infections. We are investigating the potential of bacterial communities to underlie short-term adaptation using adaptation of D. melanogaster and D. simulans to different fruit as a study system.
As the first step we profiled the diversity and composition of bacterial communities associated with Drosophila across multiple species, habitats, and substrates. We amplified and sequenced a region of the bacterial ribosomal DNA from whole body fly samples using 454 technology. We focused on comparing the bacterial communities of the sibling species D. melanogaster and D. simulans in the lab and in an ecologically and evolutionary relevant setting: their natural environment. In most cases we were able to study flies from these two species collected by aspiration from the same fruit. We also included nine different species spanning the Drosophila phylogeny to test whether phylogenetic distance and distance between bacterial communities are correlated.
We show that natural bacterial communities associated with Drosophila contain more different bacterial taxa than previously thought. Comparison to a mammalian fecal data set reveals that although mammal-associated bacterial communities are more diverse on average, the diversity of some mammalian fecal samples lies within the range or is even lower than that of the Drosophila samples we analyzed. This finding is interesting because it has been a matter of debate whether organisms with an adaptive immune system can in general accommodate higher bacterial diversity. By comparing the bacterial communities of D. melanogaster and D. simulans collected directly from different natural food substrates we demonstrate that bacterial communities differ primarily between substrates and very weakly among fly species.
We find acetic acid bacteria of the genera Acetobacter and Gluconobacter to be associated with all wild-caught flies constituting two thirds of all sequences. Acetic acid bacteria oxidize sugars and ethanol to acetic acid and are known to be directly involved in the development of a specific process of decay called ‘sour rot’ on grapes that causes wine spoilage. There is previous evidence that Drosophila is vital for the dispersal of acetic acid bacteria among rotting fruit: grapes covered with nets in the field do acquire yeasts, but no acetic acid bacteria and acetic acid bacteria thrive on grapes only when flies are present. At the same time, Acetobacter has been shown to promote Drosophila larval growth and shorten development time under certain nutritional conditions. Therefore, we argue that the relationship between Acetobacteraceae and Drosophila is likely mutualistic.
Individual natural fly samples are dominated by bacteria known to be pathogenic in Drosophila, such as Enterococcus and Providencia. These bacteria are known to reach very high cell counts during systemic infections of Drosophila and we believe that the inclusion of systemically infected flies in these samples is the most likely explanation for the observed pattern. The observation that it is in principle possible to identify potential candidate pathogens in natural populations using standard, high throughput microbial community screening techniques opens up opportunities for large scale epidemiological studies in nature and can help to identify candidate pathogenic bacterial species for further investigation in the laboratory.
In the laboratory, fly associated bacterial communities are similar irrespective of phylogenetic distance between fly species, suggesting that host genetic factors either play a minor role in shaping the bacterial communities associated with Drosophila or, as suggested by the difference of bacterial communities between D. melanogster and D. simulans in the wild, require natural conditions to manifest themselves. High variability of Drosophila bacterial communities within and between laboratories is a potential source of experimental noise when studying phenotypic variation. The impact of microbes on Drosophila phenotypes ranges from influencing growth to cold tolerance and it is hard to imagine traits that are not subject in principle to alteration by microbes.
We hope that our data will serve as a solid foundation for future studies especially for the growing community of scientists that are interested in the microbial communities that are associated with Drosophila.
Fabian Staubach and Dmitri Petrov

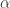 -proteobacterium Wolbachia and an unnamed
-proteobacterium Wolbachia and an unnamed 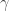 -proteobacterium from the Enterobacteriaceae. Microbial communities varied among host populations for measures of community diversity and exhibited significant population structure. We also uncovered a strong negative correlation in the abundance of the two dominant OTUs, suggesting they may fulfill similar roles as nutritional mutualists. This broad survey represents the most comprehensive assessment, to date, of the microbes that associate with bed bugs, and uncovers evidence for potential antagonism between the two dominant members of the bed bug microbiome.
-proteobacterium from the Enterobacteriaceae. Microbial communities varied among host populations for measures of community diversity and exhibited significant population structure. We also uncovered a strong negative correlation in the abundance of the two dominant OTUs, suggesting they may fulfill similar roles as nutritional mutualists. This broad survey represents the most comprehensive assessment, to date, of the microbes that associate with bed bugs, and uncovers evidence for potential antagonism between the two dominant members of the bed bug microbiome.