This guest post is by Peter Ralph and Graham Coop (@graham_coop) on their paper The geography of recent genetic ancestry across Europe arXived here
In this paper we look at the genetic traces of very recent common ancestry between pairs of individuals from across Europe. We’ll likely write a few more accessible posts on this work when the paper is closer to publication, but for now (in the spirit of Haldane’s sieve) we write a little bit more of a technical post now [the full details are in the paper].
We started this project wanting to estimate recent migration rates, across continents like Europe — if we could learn how far away distant cousins are from each other, then, all else equal, we could then estimate typical migration distances. This isn’t where we ended up (that’s another project we are working on), but the basic idea, of looking at the geographic distribution of close relatives, led us to some interesting places.
As most populations lack the amazing pedigrees like that worked on by Decode in Iceland [e.g. here] we can’t actually know the true relationships between the samples (other than a few obvious siblings and full cousins). However, long segments of chromosome shared (almost) identically by descent (IBD) between two people have probably been inherited from a recent common ancestor. The length of these IBD segments tells us something about how long ago the ancestor lived,
since the older the ancestor, the more opportunity for recombination to whittle down the segment.
This has been worked on by a bunch of different groups, but the historical inference has usually been applied to small or relatively isolated populations. To really push the boundaries of these approaches we used the European subset of the POPRES dataset, which consists of thousands of human individuals. This is currently one of the best genome-scale, geographically indexed datasets, and represents a huge outbred population where we’d expect patterns of variation to be (at least partly) due to continuous migration, rather than, say, recent mixing of diverged populations or bottlenecks. So, we ran BEAGLE on the dataset to find IBD segments, and got lots of wonderful signal — it turned out that most pairs of people in the European sample (around 75%) shared IBD segments that were megabases long (i.e. longer than 1 centi-Morgan, cM). After a bunch of power and false-positive simulations, we were convinced that most of those blocks of IBD had been inherited from single common ancestors.
You could think of our results in two pieces: first, doing descriptive statistics on the distribution of IBD abundances and lengths across geography; and second, doing some inference on this distribution to see what we can learn about when those common ancestors lived.
As we hoped, there was a nice relationship to geography – people nearer to each other typically shared more and longer IBD than people farther away, in a nice monotonic relationship. This convinced us that continuous, local migration had played an important role in shaping current patterns of relatedness across Europe. Geographic distance was definitely not the only factor – superimposed on top of this was distinctive regional variation. For example, of one the strongest signals we saw was that there are higher levels of IBD sharing in Eastern Europe. As you’ll see from the paper, after further work, we think this is a potential signal of the Slavic or Hunnic expansions.
There were also some surprises to us along the way — like people in the UK sharing more IBD with Irish than with other people in the UK — that turned out to make sense after thinking about rapidly growing populations with directional migration (although there are other explanations). We correlated the patterns we saw with historical events, but (as with most genomic studies of human history) there was a lot of uncertainty. Sure, the patterns we see are consistent with the story we told, but there could potentially be a lot of other explanations, especially given the complicated and often unknown demographic history of European populations. What if all the IBD we saw came from the Neolithic expansion rather than the last two or three thousand years? This turns out to be a bigger worry than you might think — it’s fairly unlikely that two people inherited a 3cM block from a single common ancestor from 6000 years ago,but if they have enough common ancestors from back then (e.g. a strong enough bottleneck), it turns out to be reasonably likely.
So, we did some coalescent theory to work out the relationship between numbers of shared ancestors back through time (closely related to coalescent rate) and the observed distribution of IBD block lengths. We could then invert this relationship to estimate from the observed distribution of IBD blocks the mean number of ancestors that pairs of people from different parts of Europe share with each other, as a function of time. Unfortunately, this turns out to have a lot of unavoidable uncertainty– the inversion problem is “ill-conditioned” (in other terms, the likelihood surface is ridged), meaning that there were a lot of different histories that gave the same IBD length distribution.
Despite this, we could still rigorously learn a lot of good information — in particular, nearly all the IBD blocks we found did actually come from ancestors living during the last 3,000 years. Although we could only tie down the ages of the common ancestors to within a few hundred year, the major patterns can be likely tied to known historical events. There is quite a bit of uncertainty about the specific interpretations — it is still not straightforward to go from pairwise numbers of shared ancestors 1,500 years ago to conclusions about demographic events at the time — but used in conjunction with other sources of information has the promise to conclusively resolve some longstanding debates about recent history.
Finally, two addendums (addenda?) about the methods: The first is that we took an empirical approach to estimating the relationship between coalescent time distribution and observed IBD block length, by simulating a bunch (actually copying over blocks and re-running BEAGLE). We did this because BEAGLE is effectively a black box, for our purposes. This sort of approach is more common in experimental physics, where the empirical properties of detectors have to be worked out (and the problem of inferring the signal is known as “data unfolding”).
Second, we should emphasize that the uncertainty we came across in inferring dates is theoretically unavoidable, using IBD block length data. We think this is a common issue for many sorts of population genetics data — situations in which, even though we have a ton of data, getting specific, tightly constrained inferences requires making fairly strong assumptions (or equivalently, working in a specific set of parametric models). This has been highlighted in some cases [like this one], but more work is needed on this to ensure that we represent the inherent uncertainties in population genetics inferences correctly.
We’d love feedback from the popgen community about what aspects of the paper they’d like to see clarified/improved [obviously it is a pretty involved paper already, so concentrate on specific suggestions]. We have a tonne more ideas of how to improve this inference technology and extend it to other applications. But we’d love to hear your thoughts too.


 ) across 2,930 orthologous gene alignments from 34 Escherichia coli genomes, and found substantial variation among genes in the density of synonymous polymorphisms. They argue that this pattern reflects variation in the mutation rate per nucleotide (
) across 2,930 orthologous gene alignments from 34 Escherichia coli genomes, and found substantial variation among genes in the density of synonymous polymorphisms. They argue that this pattern reflects variation in the mutation rate per nucleotide (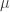 ) among genes. However, the effective population size (N) is not necessarily constant across the genome. In particular, different genes may have different histories of horizontal gene transfer (HGT), whereas Martincorena et al. used a model with random recombination to calculate
) among genes. However, the effective population size (N) is not necessarily constant across the genome. In particular, different genes may have different histories of horizontal gene transfer (HGT), whereas Martincorena et al. used a model with random recombination to calculate  . They did filter alignments in an effort to minimize the effects of HGT, but we doubt that any procedure can completely eliminate HGT among closely related genomes, such as E. coli living in the complex gut community.
. They did filter alignments in an effort to minimize the effects of HGT, but we doubt that any procedure can completely eliminate HGT among closely related genomes, such as E. coli living in the complex gut community.