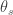Defensive complexity and the phylogenetic conservation of immune control
Erick Chastain, Rustom Antia, Carl T. Bergstrom
(Submitted on 13 Nov 2012)
One strategy for winning a coevolutionary struggle is to evolve rapidly. Most of the literature on host-pathogen coevolution focuses on this phenomenon, and looks for consequent evidence of coevolutionary arms races. An alternative strategy, less often considered in the literature, is to deter rapid evolutionary change by the opponent. To study how this can be done, we construct an evolutionary game between a controller that must process information, and an adversary that can tamper with this information processing. In this game, a species can foil its antagonist by processing information in a way that is hard for the antagonist to manipulate. We show that the structure of the information processing system induces a fitness landscape on which the adversary population evolves. Complex processing logic can carve long, deep fitness valleys that slow adaptive evolution in the adversary population. We suggest that this type of defensive complexity on the part of the vertebrate adaptive immune system may be an important element of coevolutionary dynamics between pathogens and their vertebrate hosts. Furthermore, we cite evidence that the immune control logic is phylogenetically conserved in mammalian lineages. Thus our model of defensive complexity suggests a new hypothesis for the lower rates of evolution for immune control logic compared to other immune structures.