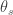The evolution of genetic architectures underlying quantitative traits
Etienne Rajon, Joshua B. Plotkin
(Submitted on 31 Oct 2012)
In the classic view introduced by R.A. Fisher, a quantitative trait is encoded by many loci with small, additive effects. Recent advances in QTL mapping have begun to elucidate the genetic architectures underlying vast numbers of phenotypes across diverse taxa, producing observations that sometimes contrast with Fisher’s blueprint. Despite these considerable empirical efforts to map the genetic determinants of traits, it remains poorly understood how the genetic architecture of a trait should evolve, or how it depends on the selection pressures on the trait. Here we develop a simple, population-genetic model for the evolution of genetic architectures. Our model predicts that traits under moderate selection should be encoded by many loci with highly variable effects, whereas traits under either weak or strong selection should be encoded by relatively few loci. We compare these theoretical predictions to qualitative trends in the genetics of human traits, and to systematic data on the genetics of gene expression levels in yeast. Our analysis provides an evolutionary explanation for broad empirical patterns in the genetic basis of traits, and it introduces a single framework that unifies the diversity of observed genetic architectures, ranging from Mendelian to Fisherian.