This guest post is by William DeWitt on his preprint (with co-authors) “Immunosequencing reveals diagnostic signatures of chronic viral infection in T cell memory”.
This is a post about our preprint at biorXiv. Although it’s a paper on infectious disease and immunology, our colleague (and Haldane’s Sieve contributor) Bryan Howie suggested we might engage the community here, since we think there are interesting connections to standard GWAS methodology. I’ll start with a one-paragraph immunology primer, then summarize what we’ve been up to, and what’s next.
Cell-mediated adaptive immunity is effected by T cells, which recognize infection through interface of the T cell receptor (TCR) with foreign peptides presented on the surface of all nucleated cells by major histocompatibility complex (MHC). During development in the thymus, maturing T cells somatically generate genes encoding the TCR according to a random process of V(D)J recombination and are passed through selective barriers against weak MHC affinity (positive selection) and strong self-peptide affinity (negative selection). This results in a diverse repertoire of self-tolerant receptors from which to deploy specific responses to threats from a protean universe of evolving pathogens. Upon recognition of foreign antigen, a T cell proliferates, generating a subpopulation with identical-by-descent TCRs. This clonal selection mechanism of immunological memory implies that the TCR repertoire dynamically encodes an individual’s pathogen exposure history, and suggests that infection with a given pathogen could be recognized by identifying concomitant TCRs.

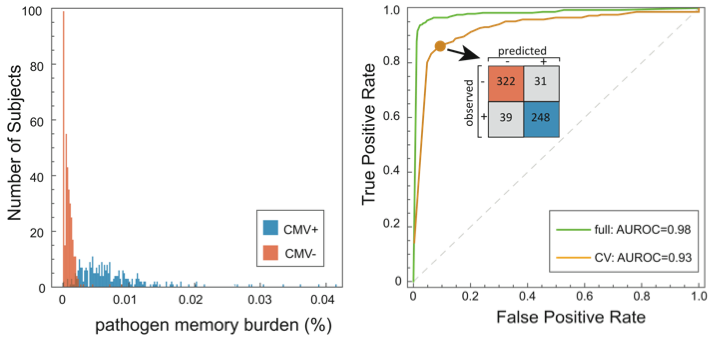
In this study, we identify signatures of viral infection in the TCR repertoire. With a cohort of 640 subjects, we performed high-throughput immunosequencing of rearranged TCR genes, and serostatus tests for cytomegalovirus (CMV) infection. We used an analysis approach similar to GWAS; among the ~85 million unique TCRs in these data, we tested for enrichment of specific TCRs among CMV seropositive subjects, identifying a set of CMV-associated TCRs. These were reduced to a single dimension by defining the pathogen memory burden as the fraction of an individual’s TCRs that are CMV-associated, revealing a powerful discriminator. A binary classifier trained against this quantity demonstrated high cross validation accuracy.

The binding of TCR to antigen is mediated by MHC, which is encoded by the highly polymorphic HLA loci. Thus, the affinity of a given TCR for a given antigen is modulated by HLA haplotype. HLA typing was performed for this cohort according to standard methods, and we investigated enrichment of specific HLA alleles among the subjects in which each CMV-associated TCR appeared. Most CMV-associated TCRs were found to have HLA restrictions, and none were associated with more than one allele in any locus.
There is substantial literature identifying TCRs that bind CMV antigen through low-throughput in vitro methods, including so-called public TCRs, which arise in many individuals. Most public CMV TCRs were present in our data, however most were not in our list of diagnostically useful CMV-associated TCRs. This is understood by considering that V(D)J recombination produces different TCRs with different probability. Public TCRs, having high recombination probability, will be repeatedly recombined in the repertoires of all subjects, regardless of CMV infection status; their presence is not diagnostic for CMV serostatus, even if they are CMV-avid. Conversely, CMV-avid TCRs with low recombination probability will be private to one subject (if present at all) in any cohort of reasonable size; their enrichment in CMV seropositive subjects is not detectable. CMV-avid TCRs with intermediate recombination probability recombine intermittently, residing transiently in the repertoires CMV naïve individuals and reliably proliferating upon CMV exposure; the presence of these TCRs in an immunosequencing sample is diagnostic for infection status.
It may be interesting to draw a comparison with GWAS, where selection drives disease-associated variants with high effect size to low population frequency, out of reach of the detection power of any study. In contrast, the V(D)J recombination machinery is constant across individuals, and CMV-avid TCRs appear to span a broad range of recombination probabilities from public to private. This includes plenty in an intermediate regime of what we might call diagnostic TCRs, which can be used to build powerful classifiers of disease status that aren’t blunted by suppression of the most relevant features.
We’ll be making the data from this study available online, constituting the largest publically accessible TCR immunosequencing data set. It’ll be fun to see what other groups do with it.
Some things we’re still working on:
• We did cross validation to assess diagnostic accuracy (yellow curve in the ROC figure), including recomputation of CMV-associated TCRs for each holdout. Results are encouraging, but a more convincing test will be to diagnose a separate cohort. We’re in the process of acquiring these data.
• MHC polymorphism suggests that thymic selection barriers censor different TCRs in individuals with different HLA haplotype. We’ve done preliminary work identifying TCRs that are associated with more common HLA alleles, indicating the possibility of HLA typing from immunosequencing data. Interestingly, this necessitates a two-tailed test due to modulation of both positive and negative selection.
• Our association analysis relies on the recurrence of TCRs with identical amino acid sequence across individuals, but we’d like to be able to define TCR motifs more loosely, so that we can detect enrichment without requiring identity. This necessitates a similarity metric in amino acid space that captures similarity in avidity. We have some ideas here, and are testing them out on some validation data. It’s definitely a tough one, but could substantially increase power in this sort of study.