[This author post is by Justin Blumenstiel and Casey Bergman on An age-of-allele test of neutrality for transposable element insertions not at equilibrium, available from the arXiv here]
Studies over the past several decades in Drosophila melanogaster have demonstrated that TE insertion alleles in natural populations tend to segregate at low frequency, particularly in regions of the genome that have a high recombination rate where natural selection is most effective. These results have largely supported a model where natural selection acts to remove deleterious TE insertions from the genome. The prevailing model of why TE insertions are deleterious is that they lead to chromosomal aberrations that occur when dispersed, non-allelic repeated sequences crossover with one another. This model is known as the ectopic recombination model and it has an important feature. Since each new insertion has the potential to recombine with all the other copies in the genome, fitness will go down faster and faster with each new copy. This yields a stable equilibrium in TE copy number.
But, are TEs at equilibrium in natural populations? Genome sequencing studies have shown that the rate of TE proliferation can vary widely over time and any given TE family may demonstrate non-equilibrium “boom and bust” behavior. How do we reconcile studies that assume equilibrium with the fact that we know TE dynamics are not at equilibrium? To deal with this problem, I began developing this model out of a class project with John Wakeley while I was a graduate student over a decade ago. This model arose of some work I published in 2002 with Hartl and Lozovsky on the age structure of non-LTR elements in D. melanogaster. I wrote this model up for my Ph.D. thesis and presented a preliminary version in a paper with Neafsey and Hartl in 2004, but it sat on the back burner until I reviewed a paper by Bergman and Bensasson in 2007 that showed many TE families in D. melanogaster have recently inserted in the genome and may not be at equilibrium.
Shortly after their paper came out I contacted Casey with the model from my thesis and we decided to push this idea forward as a collaboration, which has taken several a few years to come to fruition (both being busy with other projects and starting our labs). Things started to really move ahead when Miaomiao He in Casey’s lab generated a crucial data set that could be specifically applied to the model – strain-specific presence/absence data for a very large number of TE insertions ascertained from the D. melanogaster genome sequence. After a few more years with it on simmer, working out several kinks in the mean time (e.g. incorporating host demography, trying many different methods for estimating the posterior distribution of TE ages), Casey and I finally wrapped it up just as Haldane’s Sieve is starting to hit its stride. I expect that all my papers in the future will be pre-released on arXiv.
I could speak at length on the specific results, but I would just be saying what is already in the abstract. So, I would like to bring up three points for potential conversation.
First, what does it mean for TEs to be at transposition-selection balance when we know different TE families show a signature of “boom and bust” in genome sequences? There may be one way to reconcile this apparent problem. Any particular TE family may in fact not be at transposition-selection balance. For example, the P element, which invaded Drosophila melanogaster only a few decades ago, is hardly at transposition-selection balance. Therefore, one must be careful in using insertion frequencies for P elements to describe general TE dynamics. However, by integrating over all TE families in the genome, one may in fact reach an approximation that might be reasonable for assuming equilibrium transposition-selection balance. But one must be careful of something I call “family ascertainment bias”. Sometimes the most recently activated TEs are the ones easiest to discover and annotate because these ones are easily cloned from insertion mutations or are most frequent in genome sequences.
Second, in this paper, we derive the probability distribution for each individual TE insertion frequency based on its age. We demonstrate that this provides a method for TE insertions that are either positively or negatively selected. In the case where we show allele frequencies are less than expected (i.e. predicted to be negatively selected), many of these are copies that have zero substitutions. In principle, all of these could have inserted one generation before the reference strain was collected for genome sequencing. The inference that selection is acting against these TEs implicitly assumes either: 1) this wasn’t the case for many of these insertions, and the posterior distribution of ages is a good representation of the true age distribution, or 2) it may have been the case, but natural selection has already acted to remove slightly older TEs from the population, therefore making them absent from the genome sequence.
Third, when putting the finishing touches on our analysis of TE insertion data in North America, we ran up against the issue that nobody has yet published an explicit demographic scenario for North American populations of D. melanogaster, similar to those that have been developed by Wolfgang Stephan‘s Lab and others for European and African populations. We found one paper by Yukilevich et al (2010) from John True’s Lab that generated similar findings to the demography of European populations, which is consistent with the idea that North America populations of D. melanogaster are mainly derived from European ancestors. However, Yukilevich et al (2010) didn’t explicitly model the admixture with African populations, which is known to occur in North American populations as shown by Caracristi and Schlötterer in 2003. We were surprised that an explicit admixture scenario has not been published yet, especially since this is crucial for interpreting the data from population genomic projects like the Drosophila Genetic Reference Panel. This should be an important line of work for someone to pursue (if it isn’t being done already) and if anyone has information about this a demographic model for North American populations of D. melanogaster, we’d be keen to know more so we can see if might improve our analysis.
Justin and Casey

 ) across 2,930 orthologous gene alignments from 34 Escherichia coli genomes, and found substantial variation among genes in the density of synonymous polymorphisms. They argue that this pattern reflects variation in the mutation rate per nucleotide (
) across 2,930 orthologous gene alignments from 34 Escherichia coli genomes, and found substantial variation among genes in the density of synonymous polymorphisms. They argue that this pattern reflects variation in the mutation rate per nucleotide (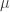 ) among genes. However, the effective population size (N) is not necessarily constant across the genome. In particular, different genes may have different histories of horizontal gene transfer (HGT), whereas Martincorena et al. used a model with random recombination to calculate
) among genes. However, the effective population size (N) is not necessarily constant across the genome. In particular, different genes may have different histories of horizontal gene transfer (HGT), whereas Martincorena et al. used a model with random recombination to calculate  . They did filter alignments in an effort to minimize the effects of HGT, but we doubt that any procedure can completely eliminate HGT among closely related genomes, such as E. coli living in the complex gut community.
. They did filter alignments in an effort to minimize the effects of HGT, but we doubt that any procedure can completely eliminate HGT among closely related genomes, such as E. coli living in the complex gut community.