
Quantitative analyses of empirical fitness landscapes
Ivan G. Szendro, Martijn F. Schenk, Jasper Franke, Joachim Krug, J. Arjan G. M. de Visser
(Submitted on 20 Feb 2012 (v1), last revised 17 Oct 2012 (this version, v2))
The concept of a fitness landscape is a powerful metaphor that offers insight into various aspects of evolutionary processes and guidance for the study of evolution. Until recently, empirical evidence on the ruggedness of these landscapes was lacking, but since it became feasible to construct all possible genotypes containing combinations of a limited set of mutations, the number of studies has grown to a point where a classification of landscapes becomes possible. The aim of this review is to identify measures of epistasis that allow a meaningful comparison of fitness landscapes and then apply them to the empirical landscapes to discern factors that affect ruggedness. The various measures of epistasis that have been proposed in the literature appear to be equivalent. Our comparison shows that the ruggedness of the empirical landscape is affected by whether the included mutations are beneficial or deleterious and by whether intra- or intergenic epistasis is involved. Finally, the empirical landscapes are compared to landscapes generated with the Rough Mt. Fuji model. Despite the simplicity of this model, it captures the features of the experimental landscapes remarkably well.
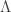 -coalescents) is provided. This recursion is used to derive asymptotic expansions as the sample size
-coalescents) is provided. This recursion is used to derive asymptotic expansions as the sample size 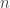 tends to infinity for the moments of the total external branch length of the Bolthausen–Sznitman coalescent. The proof is based on an elementary difference method. An alternative differential equation method is developed which can be used to obtain exact solutions for the joint moments of the external branch lengths for the Bolthausen–Sznitman coalescent. The results for example show that the lengths of two randomly chosen external branches are positively correlated for the Bolthausen–Sznitman coalescent, whereas they are negatively correlated for the Kingman coalescent provided that
tends to infinity for the moments of the total external branch length of the Bolthausen–Sznitman coalescent. The proof is based on an elementary difference method. An alternative differential equation method is developed which can be used to obtain exact solutions for the joint moments of the external branch lengths for the Bolthausen–Sznitman coalescent. The results for example show that the lengths of two randomly chosen external branches are positively correlated for the Bolthausen–Sznitman coalescent, whereas they are negatively correlated for the Kingman coalescent provided that 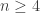 .
. 