This guest post is by Timothée Flutre and William Wen on their paper “A statistical framework for joint eQTL analysis in multiple tissues” with Matthew Stephens and Jonathan Pritchard arXived here.
As large eQTL data sets are being produced for multiple tissues, it is important to leverage all the information in the data to detect eQTLs as well as to provide ways to interpret them. Motivated by this, we developed a statistical framework for eQTL discovery that allows for joint analysis of multiple tissues. Though the details are in the paper, in this blog post we take the opportunity to highlight what we think are the main statistical features.
Looking for eQTLs in multiple tissues immediately raises the question of tissue specificity. In this paper, we define an eQTL as “active” in a particular tissue if it has a non-zero genetic effect on the expression of the target gene in this tissue. Most published works implicitly use this definition to refer to tissue-specific eQTLs. One could take issue with this definition: for example, if an eQTL is very strong in one tissue and very weak in another then one might think of this as “tissue-specific”, or at least “tissue-inconsistent”, but in our paper we stick with the binary representation of activity as a useful first step. We represent the activity pattern of a potential eQTL by a binary vector called a configuration (see Han & Eskin, PLoS Genetics 2012, and Wen & Stephens, arXiv 1111.1210). As an example, the following configuration, (110), corresponds to the case where three tissues are analyzed and the eQTL is active only in the first two tissues.
In a brief summary, we can highlight three important features of our model. First, by mapping eQTLs jointly rather than in each tissue separately, our model borrows information between the tissues in which an eQTL is active, and thereby greatly increases power. This is somewhat equivalent to relaxing the threshold of significance in the second tissue when one has already detected the eQTL in the first tissue. Second, by comparing evidence in the data for each configuration, our model provides an interpretation of how an eQTL acts in multiple tissues. In statistical terms, as more than two hypotheses are being tested (for three tissues there are 7 non-null configurations), one usually speaks of model comparison. Third, our model also estimates the proportion of each configuration in the data set. This is achieved by pooling all genes together, and thus borrowing information between them.
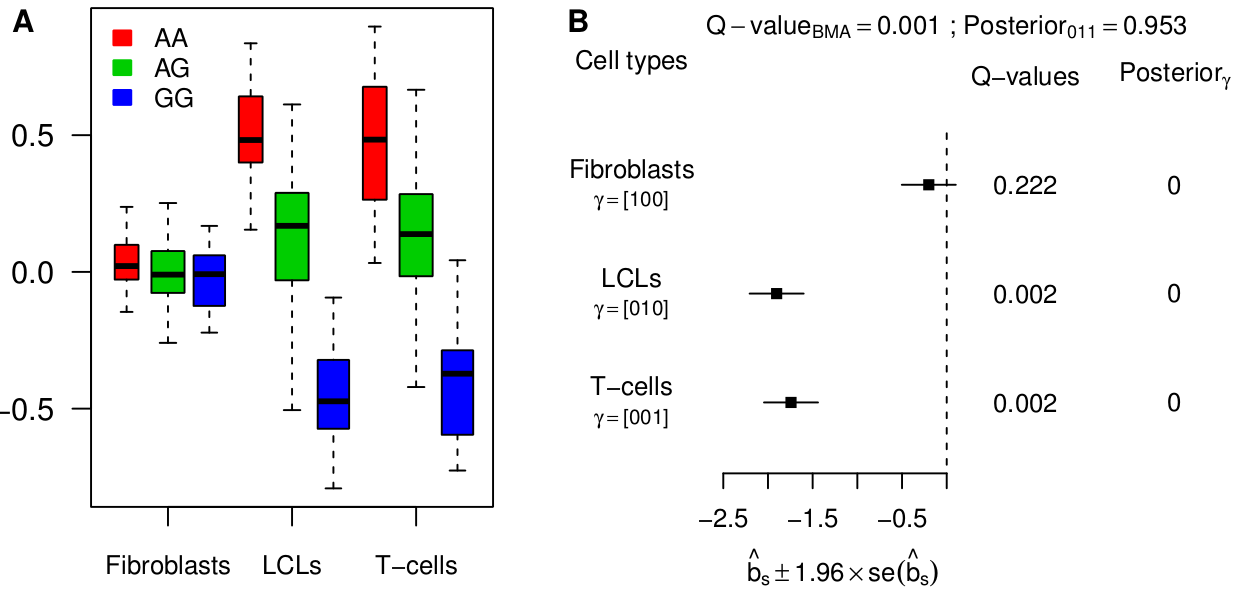
Besides simulations, we re-analyzed the largest available data set so far, 3 tissues from 75 individuals, from Dimas et al (Science 2009). Our joint analysis model has more power and detects substantially more eQTLs than a tissue-by-tissue analysis (63% at FDR=0.05). Moreover, we show how a tissue-by-tissue analysis can largely overestimate the fraction of tissue-specific eQTLs, because it does not account for incomplete power when testing in each tissue separately. Qualitatively, the discrepancy between both methods is very large on this data set. Indeed, according to the tissue-by-tissue analysis, only 19% of eQTLs are consistent across tissues, i.e. configuration (111), whereas our model estimates >80% of eQTLs to be consistent. After checking several of our assumptions, we are fairly confident in our estimate. Moreover such a high proportion of consistent eQTLs is also obtained with the pairwise approach originally used by Nica et al (2011).
The analysis of this specific data set therefore indicates that most eQTLs are consistent across tissues. Yet we find examples of strong tissue-specific eQTLs, such as between gene ENSG00000166839 (ANKDD1A) and SNP rs1628955: