
The McDonald-Kreitman Test and its Extensions under Frequent Adaptation: Problems and Solutions
Philipp W. Messer, Dmitri A. Petrov
(Submitted on 1 Nov 2012)
Population genomic studies have shown that genetic draft and background selection can profoundly affect the genome-wide patterns of molecular variation. We performed forward simulations under realistic gene-structure and selection scenarios to investigate whether such linkage effects impinge on the ability of the McDonald-Kreitman (MK) test to infer the rate of positive selection (\alpha) from polymorphism and divergence data. We find that in the presence of slightly deleterious mutations, MK estimates of \alpha\ severely underestimate the true rate of adaptation even if all polymorphisms with population frequencies under 50% are excluded. Furthermore, already under intermediate rates of adaptation, genetic draft substantially distorts the site frequency spectra at neutral and functional sites from the expectations under mutation-selection-drift balance. MK-type approaches that first infer demography from synonymous sites and then use the inferred demography to correct the estimation of \alpha\ obtain almost the correct \alpha\ in our simulations. However, these approaches typically infer a severe past population expansion although there was no such expansion in the simulations, casting doubt on the accuracy of methods that infer demography from synonymous polymorphism data. We suggest a simple asymptotic extension of the MK test that should yield accurate estimates of \alpha\ even in the presence of linkage effects.
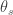
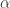 -proteobacterium Wolbachia and an unnamed
-proteobacterium Wolbachia and an unnamed 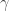 -proteobacterium from the Enterobacteriaceae. Microbial communities varied among host populations for measures of community diversity and exhibited significant population structure. We also uncovered a strong negative correlation in the abundance of the two dominant OTUs, suggesting they may fulfill similar roles as nutritional mutualists. This broad survey represents the most comprehensive assessment, to date, of the microbes that associate with bed bugs, and uncovers evidence for potential antagonism between the two dominant members of the bed bug microbiome.
-proteobacterium from the Enterobacteriaceae. Microbial communities varied among host populations for measures of community diversity and exhibited significant population structure. We also uncovered a strong negative correlation in the abundance of the two dominant OTUs, suggesting they may fulfill similar roles as nutritional mutualists. This broad survey represents the most comprehensive assessment, to date, of the microbes that associate with bed bugs, and uncovers evidence for potential antagonism between the two dominant members of the bed bug microbiome.